Preparación de muestras para el análisis bioinformático de la metilación del ADN: estrategia de asociación para la obesidad y estudios de rasgos relacionados
Summary
El presente estudio describe el flujo de trabajo para gestionar los datos de metilación del ADN obtenidos por las tecnologías de microarrays. El protocolo muestra pasos desde la preparación de la muestra hasta el análisis de datos. Todos los procedimientos se describen en detalle, y el video muestra los pasos significativos.
Abstract
La obesidad está directamente relacionada con el estilo de vida y se ha asociado con cambios en la metilación del ADN que pueden causar alteraciones en la adipogénesis y los procesos de almacenamiento de lípidos que contribuyen al desarrollo de la enfermedad. Demostramos un protocolo completo desde la selección hasta el análisis epigenético de datos de pacientes con y sin obesidad. Todos los pasos del protocolo fueron probados y validados en un estudio piloto. 32 mujeres participaron en el estudio, en el que 15 individuos fueron clasificados con obesidad según el Índice de Masa Corporal (IMC) (45,1 ± 5,4 kg/m2); y 17 individuos fueron clasificados sin obesidad según el IMC (22,6 ± 1,8 kg/m2). En el grupo con obesidad, se identificaron 564 sitios de CpG relacionados con la masa grasa mediante análisis de regresión lineal. Los sitios de CpG estaban en las regiones promotoras. El análisis diferencial encontró 470 CpGs hipometilados y 94 sitios hipermetilados en individuos con obesidad. Las vías enriquecidas más hipometiladas fueron en el RUNX, la señalización WNT y la respuesta a la hipoxia. Las vías hipermetiladas se relacionaron con la secreción de insulina, la señalización de glucagón y Ca2+. Concluimos que el protocolo identificó efectivamente los patrones de metilación del ADN y la metilación del ADN relacionada con los rasgos. Estos patrones podrían estar asociados con la expresión génica alterada, afectando la adipogénesis y el almacenamiento de lípidos. Nuestros resultados confirmaron que un estilo de vida obesogénico podría promover cambios epigenéticos en el ADN humano.
Introduction
Las tecnologías ómicas a gran escala se han utilizado cada vez más en estudios de enfermedades crónicas. Una característica interesante de estos métodos es la disponibilidad de una gran cantidad de datos generados para la comunidad científica. Por lo tanto, ha surgido una demanda para estandarizar los protocolos para permitir la comparación técnica entre estudios. El presente estudio sugiere la estandarización de un protocolo para obtener y analizar datos de metilación del ADN, utilizando un estudio piloto como ejemplo aplicado.
El gasto energético negativo predomina en los estilos de vida humanos modernos, lo que lleva a una acumulación excesiva de tejido adiposo y, en consecuencia, al desarrollo de obesidad¹. Muchos factores han aumentado las tasas de obesidad, como el sedentarismo, las dietas altas en calorías y las rutinas estresantes. La Organización Mundial de la Salud (OMS) estimó que 1.900 millones de adultos eran obesos en 2016, lo que significa que más del 20% de la población mundial tiene más de 30 kg / m2 de IMC2. La actualización más reciente de 2018 reveló que la prevalencia de obesidad en los Estados Unidos de América (EE.UU.) era superior al 42%3.
La epigenética es la adaptación estructural de las regiones cromosómicas para registrar, señalar o perpetuar estados de actividad alterados4. La metilación del ADN es una alteración química reversible en los sitios de dinucleótidos de citosina-guanosina (sitios CpG), formando 5-metilcitosina-pG (5mCpG). Puede modular la expresión génica regulando el acceso de la maquinaria de transcripción al ADN5,6,7,8. En este contexto, es esencial comprender qué sitios de CpG están asociados con rasgos relacionados con la obesidad9. Muchos factores pueden apoyar o prevenir la metilación del ADN específica del sitio. Las enzimas necesarias para este proceso, como las metiltransferasas de ADN10 (DNTM) y las translocaciones de diez-once (TETs), pueden promover la metilación o desmetilación del ADN bajo exposiciones ambientales11.
Teniendo en cuenta el creciente interés en los estudios de metilación del ADN en los últimos años, la elección de la estrategia de análisis más adecuada para responder con precisión a cada pregunta ha sido una preocupación esencial de los investigadores12,13,14. La matriz de metilación de ADN 450K es el método más popular, utilizado en más de 360 publicaciones14 para determinar el perfil de metilación de ADN. Puede determinar la metilación de hasta 485.000 CpGs localizados en el 99% de los genes conocidos15. Sin embargo, esta matriz ha sido descontinuada y reemplazada por la EPIC, cubriendo 850,000 sitios CpG. El presente protocolo se puede aplicar tanto para 450K como para EPIC16,17,18.
El protocolo se presenta paso a paso en la Figura 1 y comprende los siguientes pasos: selección de población, muestreo, preparación de experimentos, tubería de metilación de ADN y análisis bioinformático. Aquí se demuestra un estudio piloto realizado en nuestro laboratorio para ilustrar los pasos del protocolo propuesto.
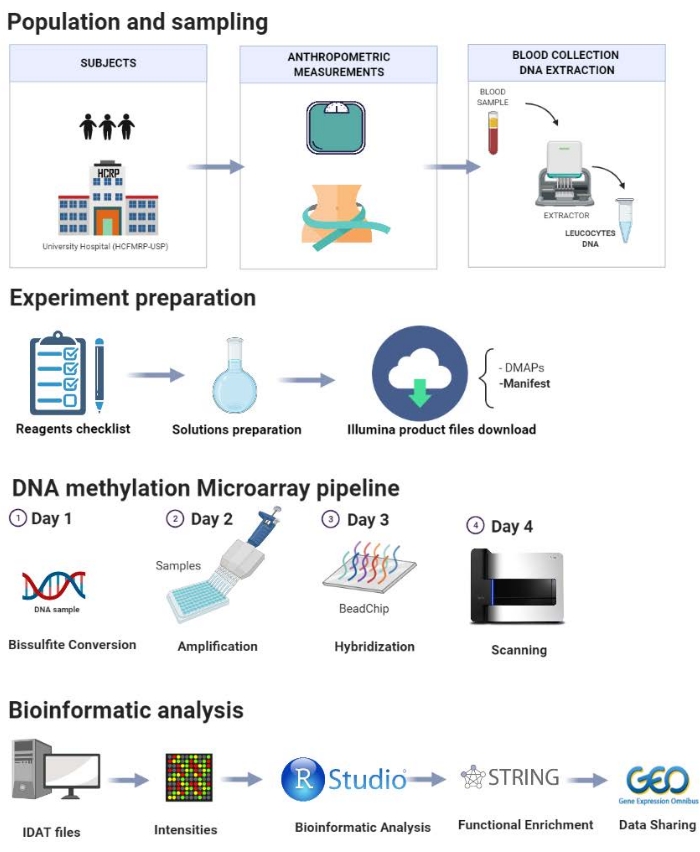
Figura 1: Esquema del protocolo presentado. Haga clic aquí para ver una versión más grande de esta figura.
Protocol
Representative Results
Discussion
Las matrices de metilación del ADN son los métodos más utilizados para acceder a la metilación del ADN debido a su relación costo-beneficio14. El presente estudio describió un protocolo detallado utilizando una plataforma de microarrays disponible comercialmente para evaluar la metilación del ADN en un estudio piloto realizado en una cohorte brasileña. Los resultados obtenidos del estudio piloto confirmaron la efectividad del protocolo. La Figura 3 muestra la comparabilidad de la muestra y la conversión completa de bisulfito32.
Como paso de control de calidad, el algoritmo ChAMP recomendó la exclusión de sitios CpGs durante el proceso de filtrado. El objetivo de excluir las sondas es mejorar el análisis de datos y eliminar el sesgo. Los CpG de baja calidad (valores de p inferiores a 0,05) se eliminaron para eliminar el ruido experimental en el conjunto de datos. Los objetivos permanecieron superados en el análisis de la gráfica de densidad. Zhou33 describió la importancia de filtrar los CpG cerca de los SNP para evitar desajustes, la mala interpretación de la metilación de las citosinas polimórficas y la causa del cambio de color del diseño de la sonda tipo I34. Además, como los cromosomas XY se ven afectados diferencialmente por la impronta, Heiss y Just35 reforzaron la importancia de filtrar esas sondas porque, en las mujeres, los problemas con la hibridación pueden ser factores de confusión35.
La fecha de vencimiento de los DMAP, la fecha de apertura de la formamida, la calidad analítica del etanol absoluto y los recuentos totales de leucocitos se consideran pasos críticos en el protocolo.
Además, según nuestras observaciones, la estimación del tipo celular es esencial para realizar el análisis bioinformático. El método Houseman realiza la estimación del tipo de célula como se describe en el estudio de Tian30. Este método se basa en 473 sitios específicos de CpG que pueden predecir los porcentajes de los tipos de células más importantes, como granulocitos, monocitos, células B y células T36. Utilizamos la función recomendada “myRefbase” del paquete ChAMP. Después de la estimación, el algoritmo ChAMP ajusta los valores beta y elimina este sesgo del conjunto de datos. Este paso es crucial en los estudios centrados en la obesidad porque esta población tiene una diferencia considerable en los glóbulos blancos debido a su estado inflamatorio crónico.
Solo cambiamos el mapa de tapa original para el sello PCR común con respecto a la modificación del método y la solución de problemas. Después de cada proceso de centrifugación, el sello se cambió por uno nuevo. No pudimos usar el sellado térmico estándar y lo adaptamos usando papel de aluminio alrededor de la placa.
Aunque los ensayos comerciales se han considerado un estándar de oro para los estudios epigenéticos, una limitación del protocolo podría ser la especificidad de los reactivos y equipos de una marca única37,38,39,40. Otra limitación es la falta de indicadores que permitan identificar el correcto avance del experimento41.
La estandarización del presente protocolo representa una gran guía para la investigación epigenética, reduciendo los errores humanos durante el proceso y permitiendo un análisis de datos exitoso y la comparabilidad entre diferentes estudios.
Según nuestros resultados, los experimentos de metilación del ADN son adecuados para estudios que comparan individuos con y sin obesidad43. Además, el análisis bioinformático propuesto proporcionó datos de alta calidad y podría considerarse en estudios a gran escala.
Utilizando el análisis de SVD, identificamos que los rasgos relacionados con la obesidad (IMC, WC y FM) influyeron en la variabilidad en los datos de metilación del ADN. Como resultado significativo, la estimación del tipo celular indica que tanto las células asesinas naturales (NK) como las células B fueron más altas en las mujeres con obesidad que en las mujeres sin obesidad (Figura 5). Los mayores recuentos de esas células podrían explicarse por el estado inflamatorio de bajo grado de estos individuos44. Observamos que los pacientes con obesidad tienen CpG hipo e hipermetilados en regiones promotoras de genes asociados con la masa grasa. La mayoría de los sitios estaban hipometilados, lo que podría estar relacionado con el aumento natural de los niveles de especies reactivas de oxígeno (ROS) en estos individuos. Esta condición de estrés oxidativo puede promover la perturbación de guanina en el sitio de dinucleótido, formando 8-hidroxi-2′-desoxiguanosina (8-OHdG), lo que resulta en un sitio de dinucleótido de 5mCp-8-OHdG y causa el reclutamiento de enzimas TET. Todos estos eventos podrían ser responsables de promover la hipometilación e hipermetilación del ADN por diferentes mecanismos de acción45.
Además, la tasa de adipogénesis parece aumentar en individuos con obesidad, con aproximadamente un 10% de células nuevas a células viejas46,47. Los aportes epigenéticos, enfatizando el ambiente obesogénico, pueden alterar las tasas de proliferación y diferenciación de las células, favoreciendo el desarrollo de la masa grasa48. Los cambios epigenéticos también pueden afectar a los programas adipogénicos, facilitando o restringiendo su desarrollo. Los factores de transcripción primarios (PPARγ o C/EBPα) o el ensamblaje de complejos multiproteicos, posicionados en regiones promotoras aguas abajo operadas por incluir o excluir enzimas modificadoras epigenéticas, regulan la expresión génica a través de hiper o hipometilación45. La vía PPARγ se ha descrito previamente para alterar la vía WNT, que tenía genes enriquecidos en este estudio. Aunque todavía se desconoce cómo se produce la señalización de WNT durante la adipogénesis, estudios recientes han reportado que podría tener un papel esencial en el metabolismo de los adipocitos, particularmente en condiciones obesogénicas49.
Disclosures
The authors have nothing to disclose.
Acknowledgements
Nos gustaría agradecer a Yuan Tian, Ph.D. (tian.yuan@ucl.ac.uk) por estar disponible para responder a todas las dudas sobre el paquete ChAMP. También agradecemos a Guilherme Telles, Msc. por su contribución tanto a los temas técnicos como científicos de este documento; hizo consideraciones importantes con respecto a la epigenética y las técnicas de captura y formato de video (guilherme.telles@usp.br). Financiamiento de consumibles: Fundación de Investigación de São Paulo (FAPESP) (#2018/24069-3) y Consejo Nacional de Desarrollo Científico y Tecnológico (CNPq: #408292/2018-0). Financiamiento personal: (FAPESP: #2014/16740-6) y Programa de Excelencia Académica de la Coordinación para el Desarrollo del Personal de Educación Superior (CAPES:88882.180020/2018-01). Los datos se pondrán a disposición del público y de forma gratuita sin restricciones. Correspondencia de dirección a NYN (correo electrónico: nataliayumi@usp.br) o CBN (correo electrónico: carla@fmrp.usp.br).
Materials
| Absolute ethanol | J.T. Baker | B5924-03 | |
| Agarose gel | Kasvi | K9-9100 | |
| Electric bioimpedance | Quantum BIA 450 Q – RJL System | ||
| Ethylenediaminetetraacetic acid (EDTA) | Corning | 46-000-CI | |
| EZ DNA Methylation-Gold kit | ZymoResearch, Irvine, CA, USA | D5001 | |
| Formamide | Sigma | F9037 | |
| FMS—Fragmentation solution | Illumina | 11203428 | Supplied Reagents |
| HumanMethylation450 BeadChip | Illumina | ||
| Maxwell Instrument | Promega, Brazil | AS4500 | |
| MA1—Multi-Sample Amplification 1 Mix | Illumina | 11202880 | Supplied Reagents |
| MicroAmp Optical Adhesive Film | Thermo Fisher Scientific | 201703982 | |
| MSM—Multi-Sample Amplification Master Mix | Illumina | 11203410 | Supplied Reagents |
| NaOH | F. MAIA | 114700 | |
| PB1—Reagent used to prepare BeadChips for hybridization | Illumina | 11291245 | Supplied Reagents |
| PB2—Humidifying buffer used during hybridization | Illumina | 11191130 | Supplied Reagents |
| 2-propanol | Emsure | 10,96,34,01,000 | |
| RA1—Resuspension, hybridization, and wash solution | Illumina | 11292441 | Supplied Reagents |
| RPM—Random Primer Mix | Illumina | 15010230 | Supplied Reagents |
| STM—Superior Two-Color Master Mix | Illumina | 11288046 | Supplied Reagents |
| TEM—Two-Color Extension Master Mix | Illumina | 11208309 | Supplied Reagents |
| Ultrapure EDTA | Invitrogen | 155576-028 | |
| 96-Well Reaction Plate with Barcode (0.1mL) | ByoSystems | 4346906 | |
| 96-Well Reaction Plate with Barcode (0.8mL) | Thermo Fisher Scientific | AB-0859 | |
| XC1—XStain BeadChip solution 1 | Illumina | 11208288 | Supplied Reagents |
| XC2—XStain BeadChip solution 2 | Illumina | 11208296 | Supplied Reagents |
| XC3—XStain BeadChip solution 3 | Illumina | 11208392 | Supplied Reagents |
| XC4—XStain BeadChip solution 4 | Illumina | 11208430 | Supplied Reagents |
References
- Manna, P., Jain, S. Obesity, oxidative stress, adipose tissue dysfunction, and the associated health risks: causes and therapeutic strategies. Metabolic Syndrome and Related Disorders. 13 (10), 423-444 (2015).
- . Obesity Available from: https://www.who.int/health-topics/obesity#tab=tab_1 (2017)
- Adult Obesity Facts. Center for Disease Control and Prevention Available from: https://www.cdc.gov/obesity/data/adult.html (2021)
- Bird, A. Perceptions of epigenetics. Nature. 396, (2007).
- Kouzarides, T. Chromatin modifications, and their function. Cell. 128 (4), 693-705 (2007).
- Berger, F. The strictest usage of the term epigenetic. Seminars in Cell & Developmental Biology. 19 (6), 525-526 (2008).
- Henikoff, S., Greally, J. M. Epigenetics, cellular memory, and gene regulation. Current Biology. 26 (14), 644-648 (2016).
- Laker, R. C., et al. Transcriptomic and epigenetic responses to short-term nutrient-exercise stress in humans. Scientific Reports. 7 (1), 1-12 (2017).
- Wahl, S., et al. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature. 541, 81-86 (2017).
- Jin, B., Robertson, K. D. DNA methyltransferases, DNA damage repair, and cancer. Epigenetic Alterations in Oncogenesis. 754, 3-29 (2013).
- Jang, H. S., Shin, W. J., Lee, J. E., Do, J. T. CpG and non-CpG methylation in epigenetic gene regulation and brain function. Genes. 8 (6), 148 (2017).
- Shen, L., Waterland, R. A. Methods of DNA methylation analysis. Current Opinion in Clinical Nutrition & Metabolic Care. 10 (5), 576-581 (2007).
- Olkhov-Mitsel, E., Bapat, B. Strategies for discovery and validation of methylated and hydroxymethylated DNA biomarkers. Cancer Medicine. 1, 237-260 (2012).
- Kurdyukov, S., Bullock, M. DNA methylation analysis: choosing the right method. Biology. 5 (1), 3 (2016).
- Yong, W. -. S., Hsu, F. -. M., Chen, P. -. Y. Profiling genome-wide DNA methylation. Epigenetics & Chromatin. 9 (1), 1-16 (2016).
- Dugué, P. A., et al. Alcohol consumption is associated with widespread changes in blood DNA methylation: Analysis of cross-sectional and longitudinal data. Addiction Biology. 1, 12855 (2021).
- Colicino, E., et al. Blood DNA methylation sites predict death risk in a longitudinal study of 12, 300 individuals. Aging. 14, 14092-14124 (2020).
- Karlsson, L., Barbaro, M., Ewing, E., Gomez-Cabrero, D., Lajic, S. Genome-wide investigation of DNA methylation in congenital adrenal hyperplasia. The Journal of Steroid Biochemistry and Molecular Biology. 201, 105699 (2020).
- Ribeiro, R. R., Guerra-Junior, G., de Azevedo Barros-Filho, A. Bone mass in schoolchildren in Brazil: the effect of racial miscegenation, pubertal stage, and socioeconomic differences. Journal of Bone and Mineral Metabolism. 27 (4), 494-501 (2009).
- Filozof, C., et al. Obesity prevalence and trends in Latin-American countries. Obesity Reviews. 2 (2), 99-106 (2001).
- Chadid, S., Kreger, B. E., Singer, M. R., Bradlee, M. L., Moore, L. L. Anthropometric measures of body fat and obesity-related cancer risk: sex-specific differences in Framingham Offspring Study adults. International Journal of Obesity. 44 (3), 601-608 (2020).
- Nicoletti, C. F., et al. DNA methylation pattern changes following a short-term hypocaloric diet in women with obesity. European Journal of Clinical Nutrition. 74 (9), 1345-1353 (2020).
- Assessing Your Weight. Centers for Disease Control and Prevention Available from: https://www.cdc.gov/healthyweight/assessing/index.html (2020)
- Giavarina, D., Lippi, G. Blood venous sample collection: Recommendations overview and a checklist to improve quality. Clinical Biochemistry. 50 (10-11), 568-573 (2017).
- Duijs, F. E., Sijen, T. A rapid and efficient method for DNA extraction from bone powder. Forensic Science International: Reports. 2, 100099 (2020).
- . Infinium HD Methylation Assay, Manual Protocol Available from: https://support.illumina.com/content/dam/illumina-support/documents/documentation/chemistry_documentation/infinium_assays/infinium_hd_methylation/infinium-hd-methylation-guide-15019519-01.pdf (2015)
- Serrano, J., Snuderl, M. Whole genome DNA methylation analysis of human glioblastoma using Illumina BeadArrays. Glioblastoma. , 31-51 (2018).
- Leti, F., Llaci, L., Malenica, I., DiStefano, J. K. Methods for CpG methylation array profiling via bisulfite conversion. Disease Gene Identification. 1706, 233-254 (2018).
- Noble, A. J., et al. A validation of Illumina EPIC array system with bisulfite-based amplicon sequencing. PeerJ. 9, 10762 (2021).
- Tian, Y., et al. ChAMP: updated methylation analysis pipeline for Illumina BeadChips. Bioinformatics. 33, 3982-3984 (2017).
- Turinsky, A. L., et al. EpigenCentral: Portal for DNA methylation data analysis and classification in rare diseases. Human Mutation. 41 (10), 1722-1733 (2020).
- . The Chip Analysis Methylation Pipeline Available from: https://www.bioconductor.org/packages/release/bioc/vignettes/ChAMP/inst/doc/ChAMP.html (2020)
- Zhou, W., Laird, P. W., Shen, H. Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Research. 45 (4), 22 (2017).
- Wanding, Z., Triche, T. J., Laird, P. W., Hui, S. SeSAMe: reducing artifactual detection of DNA methylation by Infinium BeadChips in genomic deletions. Nucleic Acids Research. 46 (20), 123 (2018).
- Heiss, J. A., Just, A. C. Improved filtering of DNA methylation microarray data by detection p values and its impact on downstream analyses. Clinical Epigenetics. 11, 15 (2019).
- Houseman, E. A., et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 13, 86 (2012).
- Valavanis, I., Sifakis, E. G., Georgiadis, P., Kyrtopoulos, S., Chatziioannou, A. A. A composite framework for the statistical analysis of epidemiological DNA methylation data with the Infinium human Methylation 450K BeadChip. IEEE Journal of Biomedical and Health Informatics. 18 (3), 817-823 (2014).
- Sun, N., Zhang, J., Zhang, C., Shi, Y., Zhao, B., Jiao, A., Chen, B. Using Illumina Infinium HumanMethylation 450K BeadChip to explore genomewide DNA methylation profiles in a human hepatocellular carcinoma cell line. Molecular Medicine Reports. 18 (5), 4446-4456 (2018).
- Moran, S., Arribas, C., Esteller, M. Validation of a DNA methylation microarray for 850,000 CpG sites of the human genome enriched in enhancer sequences. Epigenomics. 8 (3), 389-399 (2016).
- Bibikova, M., et al. High density DNA methylation array with single CpG site resolution. Genomics. 98 (4), 288-295 (2011).
- Lehne, B., et al. A coherent approach for analysis of the Illumina HumanMethylation450 BeadChip improves data quality and performance in epigenome-wide association studies. Genome Biology. 16 (1), 1-12 (2015).
- Wang, J., Zhang, H., Rezwan, F. I., Relton, C., Arshad, S. H., Holloway, J. W. Pre-adolescence DNA methylation is associated with BMI status change from pre- to post-adolescence. Clinical Epigenetics. 25, 64 (2021).
- Maugeri, A. The effects of dietary interventions on DNA methylation: implications for obesity management. International Journal of Molecular Sciences. 21, 8670 (2020).
- DeFuria, J., et al. B cells promote inflammation in obesity and type 2 diabetes through regulation of T-cell function and an inflammatory cytokine profile. Proceedings of the National Academy of Sciences. 110 (13), 5133-5138 (2013).
- Kietzmann, T., Petry, A., Shvetsova, A., Gerhold, J. M., Görlach, A. The epigenetic landscape related to reactive oxygen species formation in the cardiovascular system. British Journal of Pharmacology. 174, 1533-1554 (2017).
- Chase, K., Sharma, R. P. Epigenetic developmental programs and adipogenesis: implications for psychotropic induced obesity. Epigenetics. 8 (11), 1133-1140 (2013).
- Spalding, K. L., et al. Dynamics of fat cell turnover in humans. Nature. 453, 783-787 (2008).
- Ross, S. E., et al. Inhibition of adipogenesis by Wnt signaling. Science. 289, 950-953 (2000).
- Bagchi, D. P., et al. Wnt/β-catenin signaling regulates adipose tissue lipogenesis and adipocyte-specific loss is rigorously defended by neighboring stromal-vascular cells. Molecular Metabolism. 42, 101078 (2020).
