Une analyse fiable du poids moléculaire (MW) des protéines en solution est essentielle pour la recherche biomoléculaire1,2,3,4. L’analyse de MW informe le scientifique si la protéine correcte a été produite et si elle est appropriée pour l’usage dans l’expérimentation plus loin5,6. Comme décrit sur les sites Web des réseaux de recherche sur les protéines P4EU7 et ARBRE-Mobieu8, le contrôle de la qualité des protéines doit caractériser non seulement la pureté du produit final, mais aussi son état oligomérique, son homogénéité, son identité, sa conformation, structure, les modifications post-traduction et d’autres propriétés.
La mesure MW dans la solution non dénaturante identifie la forme de la protéine qui est présente dans un environnement aqueux, qu’il soit monomérique ou oligomérique. Alors que pour de nombreuses protéines l’objectif est de produire la forme monomérique, pour d’autres un oligomère indigène spécifique est la clé de l’activité biologique9,10,11,12. D’autres oligomères et agrégats non indigènes ne sont pas souhaitables et entraîneront des défauts dans la détermination structurelle par la cristallographie, la résonance magnétique nucléaire (RMN) ou la diffusion de rayons X à petit angle, ainsi que des artefacts ou des inexactitudes dans les analyses fonctionnelles quantifier la liaison par calorimétrie de titration isothermique ou résonance plasmon de surface2,13.
Dans le cas des biothérapeutiques tels que les anticorps monoclonaux (mAbs), l’analyse MW basée sur des solutions sert un but similaire de contrôle de la qualité et de caractérisation du produit. Les agrégats et les fragments excessifs sont indicatifs d’un produit instable qui n’est pas adapté à une utilisation humaine. Les organismes de réglementation exigent une caractérisation minutieuse, non seulement de la molécule thérapeutique, mais aussi des dégradants potentiels qui peuvent être présents dans le produit final14,15,16,17.
Certaines des méthodes les plus répandues pour analyser les protéines MW sont l’électrophores de gel de sulfate de sulfate de sodium polyacrylamide (SDS-PAGE), l’électrophorèse capillaire (CE), la PAGE indigène, la spectrométrie de masse (MS), la chromatographie d’exclusion de taille (SEC) et l’analyse l’ultracentrifugation (AUC). Parmi ceux-ci, SDS-PAGE, CE et MS ne sont pas exécutés dans l’état indigène et conduisent généralement à la dissociation des oligomères et des agrégats, donc ne sont pas appropriés pour déterminer l’oligomère indigène ou quantifier les agrégats. Bien que page indigène ne, théoriquement, conserver l’état natif, dans notre expérience, il est difficile d’optimiser pour de nombreuses protéines, et les résultats ne sont pas très fiables. L’AUC, que ce soit par vitesse de sédimentation ou équilibre de sédimentation, est quantitative et peut déterminer MW à partir des premiers principes, mais il est assez lourd, nécessitant beaucoup de travail manuel et une expertise significative dans l’interprétation des données, le temps d’expérience long et un instrument très coûteux.
La SEC analytique est une méthode quantitative et relativement robuste et simple qui sépare les macromolécules pendant le flux à travers une colonne emballée. Les principes et les applications de la SEC sont bien présentés dans plusieurs revues18,19,20 et dans le manuel “Size Exclusion Chromatography: Principles and Methods”21. Les différences de rétention sont dues à différentes quantités de temps passé à se répandre dans et hors des pores dans la phase stationnaire avant d’élavoir de la fin de la colonne. Les différences proviennent (nominalement) des tailles relatives et des coefficients de diffusion des molécules22. Une courbe d’étalonnage est construite à l’aide d’une série de molécules de référence, reliant le MW de la molécule au volume d’élution. Pour les protéines, les molécules de référence sont généralement des protéines globulaires bien élevées qui n’interagissent pas avec la colonne par charge ou résidus de surface hydrophobes. Le volume d’élution est mesuré à l’aide d’un détecteur d’absorption ultraviolet (UV). Si le coefficient d’extinction UV est connu- souvent calculé à partir de la séquence, la masse totale maximale de protéines peut également être quantite.
Notamment, l’analyse de MW par SEC repose sur deux hypothèses clés concernant les protéines à caractériser : 1) elles partagent avec les normes de référence la même conformation et le même volume spécifique (en d’autres termes, la même relation entre les propriétés de diffusion et MW) et 2) comme les normes de référence, ils n’interagissent pas avec la colonne, sauf par des propriétés stériques, ils n’adhèrent pas à l’emballage de la colonne par charge ou interactions hydrophobes. Les déviations par rapport à ces hypothèses invalident la courbe d’étalonnage et conduisent à des déterminations erronées du MW. C’est le cas pour les protéines intrinsèquement désordonnées qui ont de grands radii de Stokes en raison de leurs régions non structurées étendues23,24 ou non sphériques/linéaires assemblages oligomeric10. Les protéines glycosylated auront généralement un plus grand rayon de Stokes que la forme non-glycosylated, même quand la masse ajoutée de hydrate de carbone est prise en compte19. Les protéines membranaires stétrifiantes s’élifient différemment des protéines d’étalonnage parce que leur suppression de sec dépend de la taille totale du complexe polypeptide-détergent-lipides plutôt que de l’état oligomeric et de la masse molaire de la protéine25 ,26. La chimie des colonnes, le pH et les conditions de sel affectent tous les volumes d’élution des protéines avec des résidus de surface chargés ou hydrophobes27,28.
SEC devient beaucoup plus polyvalent et fiable pour la détermination MW lorsqu’il est combiné avec la diffusion de lumière multi-angle (MALS) et différentiel réfractive index (dRI) détecteurs3,4,11,29, 30,31,32. Un détecteur d’IRD détermine la concentration en fonction de la variation de l’indice de réfraction de la solution en raison de la présence de l’analyte. Un détecteur MALS mesure la proportion de lumière dispersée par un analyte en plusieurs angles par rapport au faisceau laser incident. Collectivement connu sous le nom SEC-MALS, cet instrumentation détermine MW indépendamment du temps d’élution puisque MW peut être calculé directement à partir des premiers principes en utilisant l’équation 1,
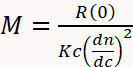 ①
①
où M est le poids moléculaire de l’analyte, R(0) le rapport réduit de Rayleigh (c.-à-d., la quantité de lumière dispersée par l’analyte par rapport à l’intensité laser) déterminée par le détecteur MALS et extrapolée à l’angle zéro, c concentration de poids déterminée par le détecteur UV ou dRI, dn/dc l’incrément de l’indice de réfraction de l’analyte (essentiellement la différence entre l’indice de réfraction de l’analyte et le tampon), et K une constante optique qui dépend de la propriétés du système telles que la longueur d’onde et l’indice de réfraction des solvants29.
Dans SEC-MALS, la colonne SEC est utilisée uniquement pour séparer les différentes espèces en solution afin qu’elles pénètrent individuellement dans les cellules du MALS et du détecteur de concentration. Le temps de rétention réel n’a aucune signification pour l’analyse, sauf dans la mesure où il résout les espèces protéiques. Les instruments sont calibrés indépendamment de la colonne et ne reposent pas sur des normes de référence. Par conséquent, SEC-MALS est considéré comme une méthode «absolue» pour la détermination MW à partir d’équations physiques de base. Si l’échantillon est hétérogène et non complètement séparé par la colonne, alors la valeur fournie à chaque volume d’élution sera une moyenne de poids des molécules dans chaque volume d’élution qui circule à travers la cellule d’écoulement par tranche de temps, environ 75 L.
En anadisant la variation angulaire de l’intensité de diffusion, MALS peut également déterminer la taille (rayon carré racine-moyen, Rg) des macromolécules et des nanoparticules avec un rayon géométrique supérieur à environ 12,5 nm29. Pour les espèces plus petites comme les protéines monomériques et les oligomères, un module dynamique de diffusion de la lumière (DLS) peut être ajouté à l’instrument MALS afin de mesurer les radii hydrodynamiques à partir de 0,5 nm et jusqu’à33.
Bien que l’analyse de la concentration des UV ou de l’IRD puisse fournir la valeur de c dans l’Eq. 1, l’utilisation de l’IRD est préférable pour deux raisons : 1) dRI est un détecteur de concentration universel, adapté à l’analyse de molécules telles que les sucres ou les polysaccharides qui ne contiennent pas d’UV chromophore34; et 2) la réponse de concentration dn/dc de presque toutes les protéines pures dans le tampon aqueux est la même à dans un ou deux pour cent (0.185 mL/g)35,ainsi il n’est pas nécessaire de connaître le coefficient d’extinction UV.
L’utilisation de SEC-MALS dans la recherche sur les protéines est assez étendue. De loin les applications les plus courantes sont d’établir si une protéine purifiée est monomeric ou oligomeric et le degré d’oligomerisation, et l’évaluation des agrégats3,10,11,17, 31,36,37,38. La capacité de le faire pour les protéines membranaires détergent-solubilisées qui ne peuvent pas être caractérisées par des moyens traditionnels est particulièrement prisée, et des protocoles détaillés pour ceci ont été édités31,39,40 , 41 Ans, états-unis ( , 42 Ans, états-unis ( , 43. D’autres applications courantes comprennent l’établissement du degré de modification post-traduction nacrée et de polydispersity de la glycoprotéine, des lipoprotéines et des conjugues similaires4,31,44 , 45 Annonces , 46 Annonces , 47; la formation (ou l’absence de celle-ci) et la stoichiométrie absolue (par opposition au rapport stoichiométrique) des hétérocomplexes, y compris les protéines-protéines, l’acide nucléique-protéine et les complexes protéique-polysaccharide24,46, 48,49,50,51,52; déterminer la dissociation d’équilibre monomère-dimer constante49,53,54; et l’évaluation de la conformation des protéines55,56. Au-delà des protéines, SEC-MALS est inestimable pour la caractérisation des peptides57,58, polymères naturels largement hétérogènes tels que les héparines59 et chitosans60,61, petits virus62 et la plupart des types de polymères synthétiques ou transformés63,64,65,66. Une bibliographie étendue peut être trouvée dans la littérature67 et en ligne (à http://www.wyatt.com/bibliography).
Ici, nous présentons un protocole standard pour l’exécution et l’analyse d’une expérience SEC-MALS. L’albumine bovine de sérum (BSA) est présentée comme exemple pour la séparation et la caractérisation des monomères et des oligomères de protéine. Le protocole BSA détermine certaines constantes du système qui servent de base à d’autres analyses SEC-MALS, y compris celles des complexes, des glycoprotéines et des protéines membranaires liées aux surfactants.
Nous notons que SEC-MALS peut être effectué en utilisant la chromatographie liquide haute performance standard (HPLC) ou l’équipement de chromatographie liquide à protéines rapides (FPLC) de nombreux fournisseurs. Ce protocole décrit l’utilisation d’un système FPLC que l’on trouve couramment dans les laboratoires qui produisent des protéines pour la recherche et le développement (voir Tableau des matériaux). Avant d’exécuter le protocole, le système FPLC, les détecteurs MALS et dRI auraient dû être installés, ainsi que leurs progiciels respectifs pour le contrôle, l’acquisition et l’analyse de données selon les instructions des fabricants et les constantes d’étalonnage requises. ou d’autres paramètres entrés dans le logiciel. Un filtre en ligne doit être placé entre la pompe et l’injecteur avec une membrane hydrophile de 0,1 m pore installée.
