El análisis fiable del peso molecular (MW) de las proteínas en solución es esencial para la investigación biomolecular1,2,3,4. El análisis MW informa al científico si se ha producido la proteínacorrecta y si es adecuada para su uso en la experimentación posterior 5,6. Como se describe en los sitios web de las redes de investigación proteica P4EU7 y ARBRE-Mobieu8, el control de la calidad de las proteínas debe caracterizar no sólo la pureza del producto final, sino también su estado oligomérico, homogeneidad, identidad, conformación, estructura, modificaciones posteriores a la traducción y otras propiedades.
La medición de MW en solución no desnaturalizante identifica la forma de la proteína que está presente en un entorno acuoso, ya sea monomérico u oligomérico. Mientras que para muchas proteínas el objetivo es producir la forma monomérica, para otros un oligómero nativo específico es clave para la actividad biológica9,10,11,12. Otros oligómeros y agregados no nativos son indeseables y darán lugar a defectos en la determinación estructural por cristalografía, resonancia magnética nuclear (RMN) o dispersión de rayos X de ángulo pequeño, así como artefactos o imprecisiones en ensayos funcionales cuantificar la unión por calorimetría de valoración isotérmica o resonancia de plasmón superficial2,13.
En el caso de bioterapias como los anticuerpos monoclonales (mAbs), el análisis de MW basado en soluciones tiene un propósito similar de control de calidad y caracterización del producto. Los agregados y fragmentos excesivos son indicativos de un producto inestable que no es adecuado para uso humano. Los organismos reguladores requieren una caracterización cuidadosa, no sólo de la molécula terapéutica, sino también de los posibles degradantes que pueden estar presentes en el producto final14,15,16,17.
Algunos de los métodos más extendidos para analizar la proteína MW son la electroforesis de gel de poliacrilamida de dodecil sulfato sódico (SDS-PAGE), la electroforesis capilar (CE), la PAGE nativa, la espectrometría de masas (MS), la cromatografía de exclusión de tamaño (SEC) y la analítica ultracentrifugación (AUC). De estos, SDS-PAGE, CE y MS no se realizan en el estado nativo y normalmente conducen a la disociación de oligómeros y agregados, por lo tanto no son adecuados para determinar el oligómero nativo o cuantificar agregados. Aunque PAGE nativo, teóricamente, conserva el estado nativo, en nuestra experiencia es difícil optimizar para muchas proteínas, y los resultados no son muy confiables. AUC, ya sea por velocidad de sedimentación o equilibrio de sedimentación, es cuantitativo y puede determinar MW a partir de los primeros principios, pero es bastante engorroso, que requiere mucha mano de obra manual y experiencia significativa en la interpretación de datos, un largo tiempo de experimento y un instrumento muy caro.
La SEC analítica es un método cuantitativo y relativamente robusto y sencillo que separa las macromoléculas durante el flujo a través de una columna empaquetada. Los principios y aplicaciones de la SEC están bien presentados en varios exámenes18,19,20 y en el manual “Cromatografía de Exclusión de Tamaño: Principios y Métodos”21. Las diferencias en la retención se deben a diferentes cantidades de tiempo dedicado a la difusión dentro y fuera de los poros en la fase estacionaria antes de eludar desde el final de la columna. Las diferencias surgen (nominalmente) de los tamaños relativos y coeficientes de difusión de las moléculas22. Una curva de calibración se construye utilizando una serie de moléculas de referencia, relacionando el MW de la molécula con el volumen de elución. En el caso de las proteínas, las moléculas de referencia son generalmente bien comportadas, proteínas globulares que no interactúan con la columna a través de la carga o residuos de superficie hidrófoba. El volumen de elución se mide con un detector de absorbancia ultravioleta (UV). Si el coeficiente de extinción UV se conoce a menudo calculado a partir de la secuencia, la masa total máxima de la proteína también puede ser cuantificada.
En particular, el análisis de MW por SEC se basa en dos supuestos clave con respecto a las proteínas a caracterizar: 1) comparten con las normas de referencia la misma conformación y volumen específico (en otras palabras, la misma relación entre las propiedades de difusión y MW) y 2) al igual que las normas de referencia, no interactúan con la columna excepto por propiedades legales,no se adhieren al embalaje de la columna por carga o interacciones hidrofóbicas. Las desviaciones de estos supuestos invalidan la curva de calibración y conducen a determinaciones erróneas de MW. Este es el caso de las proteínas intrínsecamente desordenadas que tienen grandes radios Stokes debido a sus extensas regiones no estructuradas23,24 o conjuntos oligoméricos no esféricos/lineales10. Las proteínas glicosiladas generalmente tendrán un radio Stokes más grande que la forma no glucosilada, incluso cuando se tenga en cuenta la masa de carbohidratos añadida19. Las proteínas de membrana solubibilizadas detergente eluyen de manera diferente que las proteínas de calibración porque su elución de SEC depende del tamaño total del complejo de polipéptidos-detergentes-lípidos en lugar del estado oligomérico y la masa molar de la proteína25 ,26. La química de las columnas, el pH y las condiciones de sal afectan todos los volúmenes de elución de proteínas con residuos de superficie cargados o hidrofóbicos27,28.
SEC se vuelve mucho más versátil y fiable para la determinación de MW cuando se combina con detectores de dispersión de luz multiángulo (MALS) e índice diferencial de refracción (dRI)3,4,11,29, 30,31,32. Un detector dRI determina la concentración en función del cambio en el índice de refracción de la solución debido a la presencia del analito. Un detector MALS mide la proporción de luz dispersa por un analito en múltiples ángulos en relación con el rayo láser incidente. Conocida colectivamente como SEC-MALS, esta instrumentación determina el MW independientemente del tiempo de elución, ya que el MW se puede calcular directamente a partir de los primeros principios utilizando la Ecuación 1,
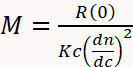 ①
①
donde M es el peso molecular del analito, R(0) la relación reducida con Rayleigh (es decir, la cantidad de luz dispersada por el analito en relación con la intensidad del láser) determinada por el detector MALS y extrapolada al ángulo cero, c el concentración de peso determinada por el detector UV o dRI, dn/dc el incremento del índice de refracción del analito (esencialmente la diferencia entre el índice de refracción del analito y el búfer), y K una constante óptica que depende de la propiedades del sistema, como la longitud de onda y el índice de refracción de disolvente29.
En SEC-MALS, la columna SEC se utiliza únicamente para separar las diversas especies en solución para que entren en las células del detector de concentración y MALS individualmente. El tiempo de retención real no tiene importancia para el análisis, excepto en cuanto a lo bien que resuelve las especies de proteínas. Los instrumentos se calibran independientemente de la columna y no se basan en normas de referencia. Por lo tanto, SEC-MALS se considera un método “absoluto” para la determinación de MW a partir de ecuaciones físicas básicas. Si la muestra es heterogénea y no está completamente separada por la columna, entonces el valor proporcionado en cada volumen de elución será un promedio de peso de las moléculas en cada volumen de elución que fluye a través de la celda de flujo por segmento de tiempo, aproximadamente 75 L.
Mediante el análisis de la variación angular de la intensidad de dispersión, MALS también puede determinar el tamaño (radio medio-cuadrado raíz, Rg) de macromoléculas y nanopartículas con un radio geométrico mayor que aproximadamente 12,5 nm29. Para especies más pequeñas como proteínas monoméricas y oligómeros, se puede añadir un módulo de dispersión de luz dinámica (DLS) al instrumento MALS para medir los radios hidrodinámicos a partir de 0,5 nm y hasta33.
Mientras que el análisis de concentración UV o dRI puede proporcionar el valor de c en Eq. 1, se prefiere el uso de dRI por dos razones: 1) dRI es un detector de concentración universal, adecuado para analizar moléculas como azúcares o polisacáridos que no contienen un UV cromóforo34; y 2) la respuesta de concentración dn/dc de casi todas las proteínas puras en tampón acuoso es la misma dentro de uno o dos por ciento (0.185 mL/g)35,por lo que no hay necesidad de conocer el coeficiente de extinción UV.
El uso de SEC-MALS en la investigación proteica es bastante extenso. Con mucho, las aplicaciones más comunes son establecer si una proteína purificada es monomérica u oligomérica y el grado de oligomerización, y evaluar los agregados3,10,11,17, 31,36,37,38. La capacidad de hacerlo para las proteínas de membrana sóloubicadas con detergente que no pueden caracterizarse por medios tradicionales es especialmente apreciada, y se han publicado protocolos detallados para ello31,39,40 , 41 , 42 , 43. Otras aplicaciones comunes incluyen el establecimiento del grado de modificación post-traduccional y polidispersidad de glicoproteína, lipoproteínas y conjugados similares4,31,44 , 45 , 46 , 47; la formación (o falta de ella) y la estequiometría absoluta (a diferencia de la relación estequiométrica) de heterocomplejos, incluidos los complejos proteína-proteína, ácido proteico-nucleico y proteína-polisacárido24,46, 48,49,50,51,52; determinar la constante de disociación monomero-dímero49,53,54; y evaluar la conformación proteica55,56. Más allá de las proteínas, SEC-MALS es invaluable para la caracterización de péptidos57,58, polímeros naturales ampliamente heterogéneos como heparinas59 y quitosanses60,61, pequeños virus62 y la mayoría de los tipos de polímeros sintéticos o procesados63,64,65,66. Una extensa bibliografía se puede encontrar en la literatura67 y en línea (a http://www.wyatt.com/bibliography).
Aquí, presentamos un protocolo estándar para ejecutar y analizar un experimento SEC-MALS. La albúmina sérica bovina (BSA) se presenta como un ejemplo para la separación y caracterización de monómeros y oligómeros proteicos. El protocolo BSA determina ciertas constantes del sistema que sirven como base para nuevos análisis SEC-MALS, incluidos los de complejos, glicoproteínas y proteínas de membrana unidas a surfactantes.
Observamos que se puede realizar SEC-MALS utilizando un equipo estándar de cromatografía líquida de alto rendimiento (HPLC) o cromatografía líquida de proteína rápida (FPLC) de muchos proveedores. Este protocolo describe el uso de un sistema FPLC que se encuentra comúnmente en laboratorios que producen proteínas para investigación y desarrollo (ver Tabla de Materiales). Antes de ejecutar el protocolo, el sistema FPLC, los detectores MALS y dRI deberían haber sido instalados, junto con sus respectivos paquetes de software para el control, la adquisición y el análisis de datos según las instrucciones del fabricante y cualquier constante de calibración requerida u otros ajustes introducidos en el software. Se debe colocar un filtro en línea entre la bomba y el inyector con una membrana hidrofílica de poros de 0,1 m instalada.
