Reliable analysis of the molecular weight (MW) of proteins in solution is essential for biomolecular research1,2,3,4. MW analysis informs the scientist if the correct protein has been produced and if it is suitable for use in further experimentation5,6. As described on the web sites of protein research networks P4EU7 and ARBRE-Mobieu8, protein quality control must characterize not only the purity of the final product, but also its oligomeric state, homogeneity, identity, conformation, structure, post-translation modifications and other properties.
MW measurement in non-denaturing solution identifies the form of the protein that is present in an aqueous environment, whether monomeric or oligomeric. While for many proteins the goal is to produce the monomeric form, for others a specific native oligomer is key to biological activity9,10,11,12. Other oligomers and non-native aggregates are undesirable and will lead to flaws in structural determination by crystallography, nuclear magnetic resonance (NMR) or small-angle X-ray scattering, as well as artifacts or inaccuracies in functional assays to quantify binding by isothermal titration calorimetry or surface plasmon resonance2,13.
In the case of biotherapeutics such as monoclonal antibodies (mAbs), solution-based MW analysis serves a similar purpose of quality control and product characterization. Excessive aggregates and fragments are indicative of an unstable product that is not suitable for human use. Regulatory agencies require careful characterization, not only of the therapeutic molecule but also potential degradants that may be present in the final product14,15,16,17.
Some of the most widespread methods for analyzing protein MW are sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), capillary electrophoresis (CE), native PAGE, mass spectrometry (MS), size-exclusion chromatography (SEC) and analytical ultracentrifugation (AUC). Of these, SDS-PAGE, CE and MS are not performed in the native state and typically lead to dissociation of oligomers and aggregates, hence are not suitable for determining the native oligomer or quantifying aggregates. Although native PAGE does, theoretically, retain the native state, in our experience it is difficult to optimize for many proteins, and results are not very reliable. AUC, whether by sedimentation velocity or sedimentation equilibrium, is quantitative and can determine MW from first principles, but it is quite cumbersome, requiring much manual labor and significant expertise in data interpretation, long experiment time and a very expensive instrument.
Analytical SEC is a quantitative and relatively robust, simple method that separates macromolecules during flow through a packed column. The principles and applications of SEC are well presented in several reviews18,19,20 and in the handbook "Size Exclusion Chromatography: Principles and Methods"21. The differences in retention are due to different amounts of time spent diffusing into and out of the pores in the stationary phase before eluting from the end of the column. The differences arise (nominally) from the relative sizes and diffusion coefficients of the molecules22. A calibration curve is constructed using a series of reference molecules, relating the MW of the molecule to elution volume. For proteins, the reference molecules are generally well-behaved, globular proteins that do not interact with the column via charge or hydrophobic surface residues. Elution volume is measured with an ultraviolet (UV) absorbance detector. If the UV extinction coefficient is known-often calculated from the sequence-the protein peak total mass may also be quantitated.
Notably, the analysis of MW by SEC relies on two key assumptions regarding the proteins to be characterized: 1) they share with the reference standards the same conformation and specific volume (in other words, the same relationship between diffusion properties and MW) and 2) like the reference standards, they do not interact with the column except by steric properties-they do not adhere to the column packing by charge or hydrophobic interactions. Deviations from these assumptions invalidate the calibration curve and lead to erroneous MW determinations. This is the case for intrinsically disordered proteins that have large Stokes radii due to their extensive unstructured regions23,24 or non-spherical/linear oligomeric assemblies10. Glycosylated proteins will generally have a larger Stokes radius than the non-glycosylated form, even when the added carbohydrate mass is taken into account19. Detergent-solubilized membrane proteins elute differently than calibration proteins because their elution from SEC depends on the total size of the polypeptide-detergent-lipids complex rather than the oligomeric state and molar mass of the protein25,26. Column chemistry, pH and salt conditions all affect elution volumes of proteins with charged or hydrophobic surface residues27,28.
SEC becomes much more versatile and reliable for MW determination when combined with multi-angle light scattering (MALS) and differential refractive index (dRI) detectors3,4,11,29,30,31,32. A dRI detector determines concentration based on the change in solution refractive index due to the presence of the analyte. A MALS detector measures the proportion of light scattered by an analyte into multiple angles relative to the incident laser beam. Collectively known as SEC-MALS, this instrumentation determines MW independently of elution time since MW can be calculated directly from first principles using Equation 1,
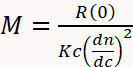 (1)
(1)
where M is the molecular weight of the analyte, R(0) the reduced Rayleigh ratio (i.e., the amount of light scattered by the analyte relative to the laser intensity) determined by the MALS detector and extrapolated to angle zero, c the weight concentration determined by the UV or dRI detector, dn/dc the refractive index increment of the analyte (essentially the difference between the refractive index of the analyte and the buffer), and K an optical constant that depends on the system properties such as wavelength and solvent refractive index29.
In SEC-MALS, the SEC column is used solely to separate the various species in solution so that they enter the MALS and concentration detector cells individually. The actual retention time has no significance for the analysis except as far as how well it resolves the protein species. The instruments are calibrated independently of the column and do not rely on reference standards. Hence, SEC-MALS is considered an 'absolute' method for MW determination from basic physical equations. If the sample is heterogeneous and not completely separated by the column, then the value provided at each elution volume will be a weight average of the molecules in each elution volume that flows through the flow cell per time slice, approximately 75 μL.
By analysis of the angular variation of scattering intensity, MALS can also determine the size (root-mean-square radius, Rg) of macromolecules and nanoparticles with geometric radius larger than about 12.5 nm29. For smaller species such as monomeric proteins and oligomers, a dynamic light scattering (DLS) module may be added to the MALS instrument in order to measure hydrodynamic radii from 0.5 nm and up33.
While either UV or dRI concentration analysis may provide the value of c in Eq. 1, use of dRI is preferred for two reasons: 1) dRI is a universal concentration detector, suitable for analyzing molecules such as sugars or polysaccharides that do not contain a UV chromophore34; and 2) the concentration response dn/dc of almost all pure proteins in aqueous buffer is the same to within one or two percent (0.185 mL/g)35, so there is no need to know the UV extinction coefficient.
The use of SEC-MALS in protein research is quite extensive. By far the most common applications are establishing whether a purified protein is monomeric or oligomeric and the degree of oligomerization, and assessing aggregates3,10,11,17,31,36,37,38. The ability to do so for detergent-solubilized membrane proteins that cannot be characterized by traditional means is especially prized, and detailed protocols for this have been published31,39,40,41,42,43. Other common applications include establishing the degree of post-translational modification and polydispersity of glycoprotein, lipoproteins and similar conjugates4,31,44,45,46,47; the formation (or lack thereof) and absolute stoichiometry (as opposed to stoichiometric ratio) of heterocomplexes including protein-protein, protein-nucleic acid and protein-polysaccharide complexes24,46,48,49,50,51,52; determining the monomer-dimer equilibrium dissociation constant49,53,54; and evaluating protein conformation55,56. Beyond proteins, SEC-MALS is invaluable for characterization of peptides57,58, broadly heterogeneous natural polymers such as heparins59 and chitosans60,61, small viruses62 and most types of synthetic or processed polymers63,64,65,66. An extensive bibliography may be found in the literature67 and online (at http://www.wyatt.com/bibliography).
Here, we present a standard protocol for running and analyzing a SEC-MALS experiment. Bovine serum albumin (BSA) is presented as an example for separation and characterization of protein monomers and oligomers. The BSA protocol determines certain system constants that serve as a foundation for further SEC-MALS analyses including those of complexes, glycoproteins and surfactant-bound membrane proteins.
We note that SEC-MALS may be performed using standard high-performance liquid chromatography (HPLC) or fast protein liquid chromatography (FPLC) equipment from many vendors. This protocol describes the use of an FPLC system commonly found in labs that produce proteins for research and development (see Table of Materials). Prior to running the protocol, the FPLC system, MALS and dRI detectors should have been installed, along with their respective software packages for control, data acquisition and analysis per manufacturers' instructions and any requisite calibration constants or other settings entered into the software. An inline filter should be placed between the pump and injector with a hydrophilic, 0.1 µm pore membrane installed.
Figure 1a,b show that three oligomeric forms of BSA: monomer, dimer and trimer, were well-separated on the 200 Å pore column with baseline resolution of monomer and dimer, while Figure 2a shows that separation on a 75 Å pore column did not achieve good monomer-dimer resolution. The latter example was included to illustrate a "poor" result; these differences in separation for the two columns may, in fact, be expected according to the manufacturer's stated separation ranges. The trimer is not fully separated from the dimer and higher oligomers are not well separated from the trimer and each other. Figure 2b is an example of noisy light scattering signal with particles present throughout the chromatogram, which precludes accurate MW determination.
Henceforth we focus on Figure 1b. The monomer, which eluted at 13.8 min, exhibits a weight-average molar mass Mw of 64.1 ± 0.4 kDa determined by MALS and hydrodynamic radius Rh of 3.54 ± 0.01 nm. These results are in agreement with the sequence mass and known hydrodynamic radius of BSA, 66.4 kDa and 3.5 nm respectively68, to within the usual accuracy of SEC-MALS, 5%3,69. The dimer, which eluted at 12 min, exhibited a Mw value of 127 ± 1 kDa determined by MALS-as expected, twice that of the monomer to within experimental precision-and Rh of 5.68 ± 0.06 nm. The trimer peak was also observed at 11 min with Mw of 194 ± 9 kDa determined by MALS, three times that of the monomer to within experimental precision, as expected. Rh of the trimer could not be determined due to low intensity of the DLS signal.
The molar mass points calculated across the monomer peak are uniform to within 2-5%, indicating homogeneity. It is not unusual to find a trailing shoulder with molar mass in the range of 38-50 kDa, corresponding to BSA fragments70. The molar mass points across the dimeric and trimeric peaks are not uniform, indicative of heterogeneity. The dimer peak is somewhat heterogeneous due to traces of trimer that bleed into the dimer peak, and the trimer peak is heterogeneous due to co-elution of poorly-resolved higher oligomers.
The signal-to-noise level of the monomer peak is quite acceptable in all three signals, over 100:1, as is the dimer peak with signal-to-noise of 40:1. Chromatogram regions beyond the protein peaks are flat, with the exceptions of a peak due to particulates near the total exclusion (void) volume in the LS trace and a (positive) salt peak and a (negative) dissolved air peak in the dRI trace, near the total permeation volume. These are pointed out in Figure 1a.
Level of purity can also be calculated in a SEC-MALS experiment: mass fraction of the monomeric peak in the report represents the percent of purity of the monomeric form. For BSA monomer, the calculated purity is 88%.
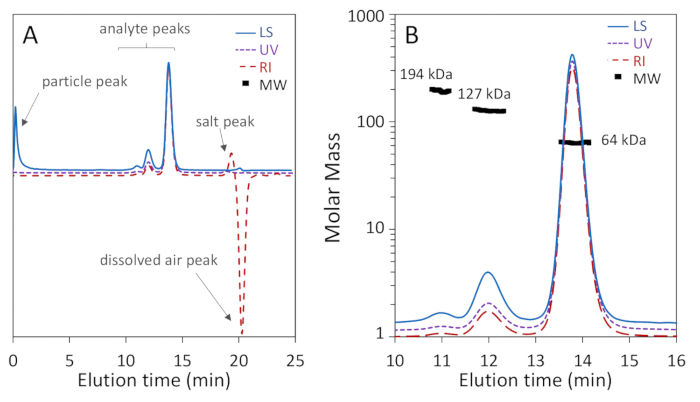
Figure 1: SEC-MALS analysis of bovine serum albumin (BSA) using a 200 Å pore size-exclusion column. Chromatogram traces are normalized to the monomer peak and offset for clarity. (A) Common artifacts that may be ignored are pointed out, including a particle peak near the beginning of the light scattering signal as well as salt and dissolved air peaks near the total permeation volume in the refractive index signal. (B) The chromatogram exhibits excellent monomer-dimer-trimer separation and the light scattering signal exhibits high signal-to-noise. The monomer and dimer MW values exhibit high homogeneity. Please click here to view a larger version of this figure.
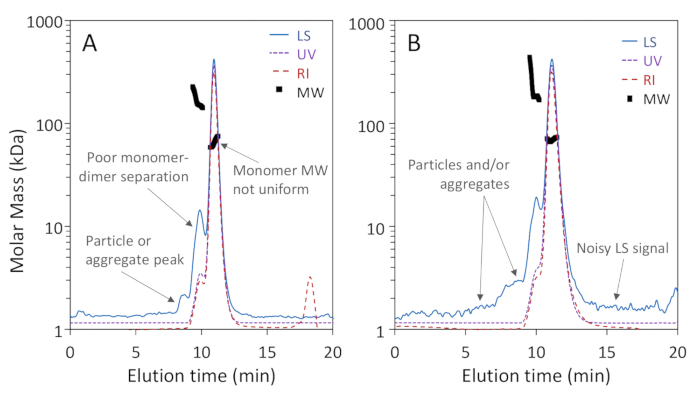
Figure 2: Examples of low-quality SEC-MALS analyses. Chromatogram traces are normalized to the monomer peak and offset for clarity. (A) Inadequate separation on a 75 Å pore size exclusion column; a particle peak between 8 – 9 min is not well separated from the proteins. (B) Inadequate signal-to-noise ratio and extensive particles adjacent to the proteins are apparent in the light scattering (LS) signal. Please click here to view a larger version of this figure.
