Antimicrobial Synergy Testing by the Inkjet Printer-assisted Automated Checkerboard Array and the Manual Time-kill Method
Summary
Antimicrobial synergy testing is used to evaluate the effect of two or more antibiotics used in combination and is typically performed by one of two methods: the checkerboard array or the time-kill assay. Here, we present an automated, inkjet printer-assisted checkerboard array synergy technique and a classic time-kill synergy study.
Abstract
As rates of multidrug-resistant (MDR) pathogens continue to rise, outpacing the development of new antimicrobials, novel approaches to treatment of MDR bacteria are increasingly becoming a necessity. One such approach is combination therapy, in which two or more antibiotics are used together to treat an infection against which one or both of the drugs may be ineffective alone. When two drugs, in combination, exert a greater than additive effect, they are considered synergistic. In vitro investigation of synergistic activity is an important first step in evaluating the possible efficacy of drug combinations. Two main in vitro synergy testing methods have been developed: the checkerboard array and the time-kill study. In this paper, we present an automated checkerboard array method that makes use of inkjet printing technology to increase the efficiency and accuracy of this technique, as well as a standard manual time-kill synergy method. The automated checkerboard array can serve as a high-throughput screening assay, while the manual time-kill study provides additional, complementary data on synergistic activity and killing.
The checkerboard array is a modification of standard minimum inhibitory concentration (MIC) testing, in which bacteria are incubated with antibiotics at different concentration combinations and evaluated for growth inhibition after overnight incubation. Manual performance of the checkerboard array requires a laborious and error-prone series of calculations and dilutions. In the automated method presented here, the calculation and dispensing of required antibiotic stock solution volumes are automated through the use of inkjet printer technology. In the time-kill synergy assay, bacteria are incubated with the antibiotics of interest, both together and individually, and sampled at intervals over the course of 24 h for quantitative culture. The results can determine whether a combination is synergistic and whether it is bactericidal, and provide data on inhibition and killing of bacteria over time.
Introduction
The spread of multidrug-resistant (MDR) bacterial pathogens, particularly MDR Gram-negative bacteria such as carbapenem-resistant Enterobacteriaceae (CRE), has left clinicians with increasingly limited options for successful anti-infective therapy1, a problem exacerbated by the sluggish pace of novel antibacterial drug discovery2,3. Antimicrobial synergy, in which two drugs used in combination exert a greater-than-additive effect, offers the possibility of salvaging existing antibiotics for use in treatment of MDR bacteria, even when these bacteria are resistant to one or both of the antibiotics individually. The techniques described in this paper provide two complementary methods of in vitro synergy testing that, when used together, allow investigators to efficiently screen antimicrobial combinations of interest for evidence of synergistic activity (the automated checkerboard array method) and then to further evaluate the kinetics of inhibition and killing demonstrated by promising combinations identified in the screening stage (the manual time-kill method).
One of the most commonly used methods of in vitro synergy testing is the checkerboard array assay, a modification of minimum inhibitory concentration (MIC) testing in which the inhibitory activity of two different antibiotics against a bacterial isolate are tested over a range of concentration combinations4,5. If the two drugs exert greater than additive activity when used together, the combination is considered synergistic6. However, setting up a checkerboard array manually involves a series of calculations and diluting and pipetting steps that are laborious and vulnerable to human error. These constraints have had the effect of limiting the use of synergy testing primarily to the retrospective evaluation of small numbers of antibiotic combinations and bacterial isolates, and results have not always been consistent among studies7,8,9,10,11. Furthermore, the complexity of synergy testing has contributed to its unavailability in the clinical microbiology laboratory and to the virtual absence of in vitro synergy testing data from clinical studies of combination therapy12,13.
In order to increase the efficiency and throughput of the checkerboard array method, we made use of an automated MIC testing technique previously developed in our laboratory that uses inkjet printing technology to precisely and consistently dispense small volumes of antibiotic stock solution into wells in a microtiter plate14. The platform obviates the need for complex calculations and multiple pipetting steps. The associated software calculates and dispenses appropriate volumes of antibiotics to create a two-dimensional checkerboard array if the user simply inputs the desired concentration range and stock solution concentration of the antibiotics. We initially tested this method against a collection of CRE isolates15 and subsequently have focused on testing colistin-containing combinations for activity against colistin-resistant isolates16. Colistin is a drug of last resort generally reserved for use in the treatment of MDR Gram-negative pathogens17,18, and colistin resistance renders already MDR bacteria nearly pan-resistant19, making them ideal candidates for the development of novel therapeutic strategies using drugs to which they are insensitive individually. We found that the combination of colistin and the protein synthesis inhibitor antibiotic minocycline had a very high rate of synergy, even against strains that were resistant to each of these drugs individually, presumably because colistin exerts a subinhibitory permeabilizing effect on even colistin-resistant bacteria. We have chosen this combination to use as an example in this paper. Of note, synergy testing can also be used to evaluate for enhanced efficacy of two drugs which are both effective individually.
The automated checkerboard array method facilitates rapid, high-throughput synergy testing. However, the checkerboard array method does have limitations. As a modified MIC assay, it provides data only on inhibition of bacterial growth and not on killing, and it does not provide data on antibiotic effects over time. By contrast, manual performance of time-kill synergy assays is more labor intensive but provides information on both inhibition and killing over a 24 h time course20,21. We used time-kill analysis on a smaller number of isolates to confirm our checkerboard array results and to determine whether the synergistic combinations we identified were also bactericidal.
Both checkerboard array and time-kill synergy methods provide valuable information on the activity of drug combinations, and are particularly useful in evaluating potential novel therapeutic options for highly resistant bacterial pathogens. The methods also have inherent limitations. The standard microbroth dilution MIC method has a known expected error range of 1 two-fold dilution22, which is increased when two drugs are tested together in a checkerboard array. The standard definition of synergy, which considers a combination synergistic only if the drugs are active together at one-fourth their respective MICs6, takes into account this expected variability, but such variability (which is thought to result from a combination of biological and technical fluctuations23) inevitably generates uncertainly about the reliability of synergy results. The lack of established quality-control standards for synergy testing is also a current limitation. Perhaps the most significant limitation of all synergy testing methods is the lack of established correlations between in vitro results and clinical outcomes when combinations are used to treat patients24. Simpler and more rapid synergy testing methods, such as the automated checkerboard array method described here, may facilitate the integration of in vitro synergy testing within clinical trials or other evaluations of patient outcomes in order to better characterize the relationship between in vitro and in vivo effects in the future.
The automated checkerboard array method that we present here offers an option for high-throughput screening of a variety of combinations and allows for quick evaluation of unusual, "high risk-high reward" combinations without a major investment of time and resources. The time-kill method, which we subsequently demonstrate, can provide additional supportive information on the synergistic activity of the combination and can help to characterize its bactericidal activity and antibacterial kinetics.
Protocol
CAUTION: Use appropriate safety procedures when working with bacteria. Wear gloves and a lab coat at all times. Perform work in a biosafety cabinet if aerosols will be generated or working with high risk pathogens.
NOTE: Twenty to 24 h before starting experiments, streak out the bacterial isolate(s) to be tested (from a colony-purified, minimally passaged stock frozen at -80 °C in tryptic soy broth with 50% glycerol stock) onto a blood agar plate. Incubate the plate at 35 °C in ambient air.
1. Inkjet Printer-assisted Automated Checkerboard Array Synergy
- Make antimicrobial stock solutions (colistin and minocycline).
- Determine antibiotic stock solution concentrations based on solubility of antibiotics and desired final concentrations in checkerboard array. Make 10 mg/mL stocks of colistin and minocycline for this example. Use the CLSI M100 document to determine appropriate solvents for each antibiotic25. Both colistin and minocycline are water-soluble; because the D300 inkjet printer requires the addition of surfactant for proper aqueous fluid handling, dissolve the antibiotics in ultrapure deionized water with 0.3% polysorbate 20.
- Weigh out antibiotic powder using an analytical balance and calculate volume of solvent needed to obtain goal stock concentration.
- If the antibiotic is supplied as a salt (e.g. colistin sulfate, minocycline hydrochloride) or in hydrated form (e.g. meropenem trihydrate), or if it is reported by the manufacturer to have less than 100% purity, perform a potency calculation26 to determine the quantity of solvent required.
- Follow this example of minocycline hydrochloride with a stated purity of 900 μg/mg:
Assay purity: 900 μg/mg
Water content: None
Active fraction: 0.926 (obtained by dividing the molecular weight of minocycline (457.48 Da) by the molecular weight of minocycline hydrochloride (493.94 Da)).
Potency = (Assay purity) * (1 – water content) * (active fraction)
= (900 μg/mg) * (1) * (0.926) = 833.4 μg/mg or 83.34%
Then determine the volume of solvent required as follows:
Volume (mL) = [Weight (mg) * Potency (μg/mg)] ÷ [Concentration (μg/mL)]
So, for example, if 34.7 mg of minocycline hydrochloride powder is weighed out, use the following calculation to determine the volume of solvent required to make a 10 mg/mL solution:
Volume = (34.7 mg) * (833.4 μg/mg) = 2.89 mL
10,000 μg/mL
- Pour antibiotic powder into a 15 mL conical tube and add the appropriate volume of water plus 0.3% polysorbate 20. Vortex until dissolved.
- Aliquot antibiotic stock solution into 0.5 mL microcentrifuge tubes and store at -80 °C until ready for use.
- Perform quality control (QC) of antimicrobial stocks for use in checkerboard array experiments at least one day prior to synergy testing so that QC results can be evaluated before using the stock for synergy testing.
NOTE: The QC technique described here is identical to the technique that would be used for minimum inhibitory concentration (MIC) testing of individual drugs and can be used as such with any strains of interest to the investigator.- Prepare bacterial suspension.
- Take an aliquot of each antibiotic stock out of the -80 °C freezer to start thawing while preparing bacterial suspension. Vortex once thawed to ensure that the antibiotic is in solution.
- Select an appropriate QC strain and determine the acceptable MIC range for drugs being tested based on Table 5A-1 in CLSI M10025. For the drugs here, use E. coli ATCC 25922; the MIC ranges for this strain are 0.25-1 μg/mL for minocycline and 0.25-2 μg/mL for colistin.
- Select a range of antibiotic concentrations to test that will include the entire QC range. Use the range of 0.0156 μg/mL to 8 μg/mL for minocycline and colistin for ATCC 25922.
- Add 1 mL of 0.9% sodium chloride to a 12 mm x 75 mm round bottom glass culture tube. Select one or two colonies from an overnight plate of ATCC 25922 and vortex gently to suspend.
- Check the concentration of bacteria using a McFarland reader. Adjust as needed by adding more 0.9% sodium chloride or more bacteria to achieve a 0.5 McFarland turbidity reading.
- Make a 1:300 dilution of the 0.5 McFarland suspension by adding 100 μL of the suspension to 30 mL of cation-adjusted Mueller-Hinton broth (CAMHB) in a 50 mL conical tube to reach a final cell density of 5×105 CFU/mL, as recommended by CLSI27.
- Using a sterile inoculating loop, isolation streak a drop of the starting inoculum onto a blood agar plate to confirm inoculum purity, and incubate at 35 °C in ambient air.
- Add antimicrobials to a flat-bottom, square-well, clear, untreated 384-well plate using the D300. Perform this step immediately after preparing bacterial suspension so that suspension can be added to the plates within 15 min of preparation26.
- Turn on the D300 inkjet printer and the associated computer. Open the software program.
- Start a new file. Above the image of the plate grid, right-click on Plate 1 and choose Edit plate. Select the appropriate plate type (384 well) and additional volume (50 μL).
- Add fluids (i.e., antibiotic stocks) to the protocol by clicking the plus sign next to Fluids on the left-hand panel. Add two fluids (colistin and minocycline).
- Hover over the panel that appeared for Fluid 1 and click the pencil to edit. Name the fluid “Colistin”, change Class to “Aqueous + Tween 20”, change Concentration to 10,000, and change concentration units to μg/mL (note that stock concentration is 10 μg/mL, i.e., 10,000 mg/mL). Leave Dispense by at Concentration and leave the remainder of the fields at their default settings. Click OK.
- Repeat the procedure above for Fluid 2 (minocycline).
- Click the Current Protocol tab at the top of the screen and change Concentration (mass) to μg/mL to determine the units used for final well antibiotic concentrations.
- Select 10 wells in the grid by clicking and dragging, then click Titration at the top of the screen. For Specify titration using select Highest concentration, for Fluid choose Colistin, for Highest Concentration enter 8 (make sure units are μg/mL), and for Distribution select 1:2 (50%). Leave default values in place for the rest of the window and click OK.
- Repeat the procedure above for minocycline to generate the minocycline titration.
- Save the protocol, and then click the Run button at the top left.
- Click the Start button. Load a 384-well plate (with lid removed) into the plate holder and press Loaded under the “Load Plate 1 – Synergy” prompt.
NOTE: This prompt is chosen by the software and does not indicate that synergy testing is being performed. Place a T8+ cassette into the cassette slot and press Loaded under the Load a T8+ cassette prompt. - When prompted, add antibiotic stock solution to the indicated reservoirs on the cassette. Follow instructions on the screen for proper loading and dispense carefully to avoid getting any bubbles in the solution. After each solution is added, press the Filled button.
- Once the inkjet printer has added antibiotic stock in appropriate volumes to each well and the Run completed box appears, click Close, remove the plate, and turn off the D300.
- Add bacterial suspensions to 384-well plate and incubate plate.
- Pour the previously prepared bacterial suspension into a sterile reagent reservoir.
- Use a multichannel pipette to add 50 μL of bacterial suspension to all antibiotic-containing wells. Add 50 μL of CAMHB without bacteria to an empty well; this will be the negative control well to confirm sterility of the media.
- Place plate in a 35 °C ambient air incubator and incubate for 16-20 h26.
NOTE: A different duration of incubation may be required if organisms other than Enterobacteriaceae are being tested; consult CLSI M10025 for organism-specific recommendations.
- Read plate on a microplate reader at an optical density of 600 (OD600) and analyze results.
- Using a spreadsheet program, shade cells with an OD600 value of ≥0.07 green, indicating growth, and cells with a value of <0.07 red, indicating no growth.
NOTE: These values were determined based on visual inspection of growth vs. no growth and correlation with OD readings for these experiments; OD600 readings from wells containing media alone were consistently below 0.07. Appropriate cutoffs may differ with different plate readers and bacteria. - Determine the MIC for each drug. The MIC is the lowest concentration of drug at which bacterial growth is inhibited. If the MIC is within the expected QC range according to the CLSI M100 document25, the stock solution is appropriate for use.
- Using a spreadsheet program, shade cells with an OD600 value of ≥0.07 green, indicating growth, and cells with a value of <0.07 red, indicating no growth.
- Prepare bacterial suspension.
- Prepare bacterial suspension for checkerboard array.
- Take an aliquot of each antibiotic stock out of the -80 °C freezer to start thawing while preparing bacterial suspension. Vortex once thawed to ensure antibiotic is in solution.
- Add ~1 mL of 0.9% sodium chloride to a 12 mm x 75 mm round bottom glass culture tube. Select one or two colonies from an overnight plate of bacteria (in this case, E. coli strain FDA-CDC 0494) and vortex gently to suspend these in the 0.9% sodium chloride.
- Check the concentration of bacteria using a McFarland reader. Adjust as needed by adding more 0.9% sodium chloride or more bacteria to achieve a 0.5 McFarland turbidity reading.
- Make a 1:300 dilution of the 0.5 McFarland suspension by adding 100 μL of the suspension to 30 mL of CAMHB in a 50 mL conical tube to reach a final cell density of 5 x 105 CFU/mL27.
- Add antimicrobials to a flat-bottom, square-well, clear, untreated 384-well plate using the D300.
NOTE: Perform this step immediately after preparing bacterial suspension so that suspension can be added to the plates within 15 minutes of preparation26.- Turn on the inkjet printer, start a new file, and add fluids to the protocol as in steps 1.2.2.1-1.2.2.3.
- Generate the synergy grid.
- Click the Synergy icon at the top of the screen and proceed through the steps. For Type select Two or more fluids factored together. For Plate, it is not necessary to exclude any wells; click Next on this step without making changes.
- On the Titration tab, enter the antibiotic concentrations and placement.
- In order to add minocycline in decreasing doubling dilutions from 32 to 0.031 μg/mL, in addition to a negative well with no antibiotic, down the y axis, enter 12 for Titration levels on the left panel. For Specify titration using, select Highest concentration; for Fluid, select Minocycline; for Highest concentration, enter 32 (make sure the unit is set at μg/mL; if it is not, close the Synergy dialog box and change under the Current Protocol tab). Make sure that the Include 0 value box is checked. Change Distribution to 1:2 (50%).
- Repeat these steps for colistin on the right-hand panel, using 12 titration levels and a highest concentration of 16. Click Next.
- On the Layout tab, choose Titration levels of first 2 fluids determine the number of rows and columns in a layout grid. Click Next. If the grid appears as expected, click Finish.
- Save the protocol, then click the Run button at the top left.
- Click the Start button. Load a 384-well plate (with the lid removed) into the plate holder and press Loaded under the Load Plate 1 – Synergy prompt. Place a T8+ cassette into the cassette slot and press Loaded under the Load a T8+ cassette prompt.
- When prompted, add antibiotic stock solution to the indicated reservoirs on the cassette. Follow instructions on the screen for proper loading and dispense carefully to avoid getting any bubbles in the solution. After each solution is added, press the Filled button.
- Once the D300 dispenser has added antibiotic stock in appropriate volumes to each well and the Run completed box appears, click Exit, remove the plate, and turn off the D300.
- Add bacterial suspensions to 384-well plate and incubate plate.
- Pour the previously prepared bacterial suspension into a sterile reagent reservoir.
- Use a multichannel pipette to add 50 μL of the suspension to all wells in the checkerboard array. Add 50 μL of CAMHB without bacteria to an empty well; this will be the negative control well to confirm sterility of the media. Incubate in a 35 °C ambient air incubator for 16-20 h26.
- Read plate on a microplate reader at OD600 and analyze checkerboard array results.
- First, check the purity plate and ensure that the isolated colonies are of a single morphology that is consistent with the expected morphology of the organism being tested.
- Using a spreadsheet program, shade cells to indicate growth and no growth as in step 1.2.4.1.
- Determine the MIC for each drug. For a drug that does not inhibit bacterial growth at the highest concentration tested, the MIC is considered to be off-scale.
- For each well in which growth is inhibited, determine the fractional inhibitory concentration (FIC) for each antibiotic based on that antibiotic’s MIC (see Figure 1B and Figure 2B).
NOTE: The FIC is the ratio of the concentration of antibiotic in a well in which growth is inhibited to its MIC; so for a drug with an MIC of 8 μg/mL, a well containing 8 μg/mL of that drug has an FIC of 1, while a well containing 4 μg/mL has an FIC of 0.5. - Calculate the fractional inhibitory concentration index (FICI) value for each well in which growth is inhibited as the sum of the FICs of each of the drugs in that well.
- Determine the lowest FICI at which growth is inhibited (minimum FICI). If the minimum FICI is £0.5, consider the combination synergistic; if 0.5-4, consider the combination indifferent; and if >4, consider the combination antagonistic. If the combination is synergistic at some concentration combinations but antagonistic at others, note this result but consider the combination overall antagonistic.
2. Time-kill Synergy Testing
- Make antimicrobial stock solutions. If this step is performed ahead of the experiment, freeze stocks at -80 °C until ready for use.
- Determine antibiotic stock solution concentrations based on solubility of antibiotics and desired final concentrations in time-kill studies. In this example, make colistin and minocycline stocks at a concentration of 1 mg/mL. Use the CLSI M100 document to determine appropriate solvents for each antibiotic25. Use water, as recommended, for both colistin and minocycline.
- Weigh out antibiotic powder using analytical balance and calculate volume of solvent needed to obtain goal stock concentration. If needed, perform a potency calculation prior to determining the quantity of solvent required, as described in step 1.1.2.1 above.
- Pour antibiotic powder into 15 mL conical tubes and add the appropriate volume of water. Vortex until dissolved.
- Using the manual broth microdilution method described in CLSI M0726, perform QC of antimicrobial stocks for use in time kill synergy experiments. Perform this step at least one day prior to synergy testing so that QC results can be reviewed before using the stock.
- Select an appropriate QC strain and determine the acceptable MIC range for drugs being tested based on Table 5A-1 in CLSI M10025. For the drugs here, use E. coli ATCC 25922; the MIC ranges for this strain are 0.25-1 μg/mL for minocycline and 0.25-2 μg/mL for colistin.
- Prepare antibiotic-containing broth microdilution plates.
- Select the highest concentration of antibiotic to be tested so the entire QC range can be included. Use a range of 0.016 to 8 μg/mL for minocycline and colistin for ATCC 25922.
- Dilute the antibiotic stocks to a working solution in CAMHB at two times the highest concentration needed (because it will be diluted 1:1 with the suspension of bacteria). In this example, dilute both stocks from 10 mg/mL to 16 μg/mL.
- Using a multi-channel pipette, add 100 μL of each of the 2x antibiotic suspensions to a well in the first column of a clear, round-bottom, untreated 96-well plate and add 50 μL of plain broth (i.e., without antibiotic) to each well of the subsequent columns.
- Remove 50 μL of antibiotic-containing broth from each well in the first column and add to the wells in the second column. Pipette up and down several times to mix the contents, generating an antibiotic concentration half that of the concentration in the first column.
- Repeat step 2.2.2.4 with each column, so that a series of serial two-fold dilutions, each with a volume of 50 μL, is prepared. Change pipette tips between each dilution step if desired to eliminate the possibility of antibiotic carryover. Note that the resultant concentrations are all still 2x the final concentrations, as they will subsequently be diluted 1:1 with bacterial suspension.
- Do not add any antibiotic to the final two columns, as these will be the negative and growth control columns.
- Prepare bacterial suspension.
- Prepare a 0.5 McFarland suspension from an overnight plate of E. coli ATCC 25922 in 0.9% sodium chloride as described in steps 1.2.1.5-1.2.1.6.
- Make a 1:150 dilution of the 0.5 McFarland suspension by adding 50 μL of the suspension to 7.5 mL of CAMHB.
NOTE: The final bacterial suspension will be diluted 1:300 once it is mixed 1:1 with the antibiotic solution, reaching the CLSI-recommended cell density of 5 x 105 CFU/mL27).
- Add bacteria to the microplate and incubate.
- Add 50 μL of the bacterial suspension to each well, except in the 11th column. Add 50 μL of CAMHB to the 11th column (negative control column).
- Incubate plate at 35 °C for 16-20 h.
NOTE: A different duration of incubation may be required if organisms other than Enterobacteriaceae are being tested; consult CLSI M10025 for organism-specific recommendations. - Read plate for growth visually using transmitted light and, for each antibiotic, determine the lowest concentration in which there is no growth; this is the MIC. Consult the CLSI M07 document for additional details on visual interpretation of MIC26. If the MIC is within the expected QC range, the stock solution is appropriate for use.
- Start initial culture.
- Make a 0.5 McFarland suspension of test organism in sterile 0.9% NaCl as described above.
- Add 100 μL of the 0.5 McFarland suspension to 5 mL of CAMHB in a 25 mm x 150 mm glass round bottom culture tube with stainless steel closure and vortex gently to mix.
- Using a sterile inoculating loop, isolation streak a drop of the diluted suspension onto a blood agar plate to confirm inoculum purity and incubate at 35 °C in ambient air.
- Replace closure on tube and incubate in a test tube rack on a shaker at 35 °C in ambient air for at least 3 h, until logarithmic-phase growth is reached (see step 2.6.1). Proceed to step 2.4 while the culture is in the incubator.
- Prepare antimicrobial solutions in 25 mm x 150 mm glass culture tubes.
- Take out antimicrobial stock aliquots from -80 °C freezer to thaw. Vortex once thawed to ensure antibiotic is in solution.
- While initial culture is incubating, add 10 mL of CAMHB to five autoclaved 25 mm x 150 mm glass culture tubes and add antimicrobial stock solutions as follows.
NOTE: For a synergy study, at least one drug should be at a concentration that does not affect the growth curve individually28; this can be determined by evaluating the effects of individual drug concentrations prior to the synergy study.- Tube 1: Add appropriate quantity of antibiotic #1 to obtain target final antibiotic concentration. In this case, add 10 μL of 1 mg/mL colistin stock to obtain a final colistin concentration of 1 μg/mL, as this is a concentration that is ineffective against the strain used in this example.
- Tube 2: Add appropriate quantity of antibiotic #2 to obtain final antibiotic concentration to be tested. In this case, add 10 μL of 1 mg/mL minocycline stock to obtain a final concentration of 1 μg/mL, a concentration that is ineffective against the strain being used in this example.
- Tube 3: Add the same quantity of antibiotic #1 and antibiotic #2 as used in tubes 1 and 2. In this case, add 10 μL of 1 mg/mL minocycline stock and 10 μL of 1 mg/mL colistin stock.
- Tube 4: Add no antibiotics; this will be the growth control tube.
- Tube 5: Add no antibiotics; this will be the negative control tube.
- Prepare 96 deep well polypropylene plates with 2 mL wells for serial dilutions by adding 900 µL of sterile 0.9% sodium chloride to rows B-H of columns 1-5 with a multichannel pipette.
- Prepare starting inoculum and add to tubes.
- Once the initial culture has reached logarithmic growth phase (~3 h for Klebsiella pneumoniae, the organism used in this example), remove the culture tube from the shaker, vortex gently, transfer ~1 mL of suspension to a 12 mm x 75 mm glass culture tube, and check density with a McFarland reader.
- If it is less than 1.0 McFarland, return tube to the shaker and incubate longer. If it is greater than 1.0 McFarland, add CAMHB to the tube, vortex gently, and re-sample, repeating the process until the suspension is at 1.0 McFarland.
- Add 100 μL of the 1.0 McFarland suspension to tubes 1-4 and vortex gently.
- Once the initial culture has reached logarithmic growth phase (~3 h for Klebsiella pneumoniae, the organism used in this example), remove the culture tube from the shaker, vortex gently, transfer ~1 mL of suspension to a 12 mm x 75 mm glass culture tube, and check density with a McFarland reader.
- Sample aliquots from each culture and perform serial ten-fold dilutions.
- At time 0 (immediately after adding bacteria to the tubes) and at 1, 2, 4, 6, and 24 h, remove a 150 μL aliquot from each culture tube by tilting the tube so that only the sterile pipette tip enters the tube and not the unsterile pipettor shaft during aliquot withdrawal. Add aliquots, respectively, to consecutive wells in the first row of the previously prepared 96 deep well plate. Return tubes to a test tube rack on a shaker in a 35 °C ambient air incubator immediately after removing aliquots at each time point.
- Using a multichannel pipette, remove 100 μL from row A, add to row B (which contains 900 μL of 0.9% sodium chloride), and pipette up and down 4-5 times to mix, creating a 1:10 dilution. Discard tips following each dilution step to prevent carryover of bacteria, which can lead to falsely elevated colony counts.
- Repeat step 2.7.2 for rows B-H with new pipette tips for each row.
- Plate diluted samples for colony counts using the drop plate method29,30.
- Label Mueller-Hinton agar plates with the antibiotic conditions and dilution to be plated.
- Using a multichannel pipette and extra-long tips (to ensure that tips reach into suspension), remove 10 μL from each well in column one and dispense carefully in a row onto the appropriately labeled plate. If small (100 mm diameter) plates are used, dispense 3 rows (each consisting of drops from rows A-H of a single column) per plate; on large (150 mm diameter) plates, dispense 8 rows per plate.
- Allow drops to dry completely (~15 min).
- At 24 h, place a 10 μL drop taken directly from the negative control tube in an indicated area of one of the plates to test for sterility. Invert plates and incubate overnight at 35 °C in ambient air.
- Count colonies and calculate cell density. Mark colonies with a fine-tip permanent marker on the reverse of the plate to avoid double-counting or missing colonies.
- First, check the purity plate and ensure that the isolated colonies are of a single morphology that is consistent with the expected morphology of the organism being tested.
- For each dilution series, identify drops with 3-30 colonies (typically one drop per dilution series). Count the colonies in these drops and record the count along with the dilution factor.
- If there are no drops in a dilution series with 3-30 colonies, count the colonies in the last drop with >30 colonies and the first drop with <3 colonies (these should be adjacent drops).
- For each dilution series, calculate number of colony forming units per milliliter (CFU/mL) in the sample based on the number of colonies in the drop using the following formula: CFU/mL = n(1/d)(100) where n is the number of colonies, d is the dilution factor (1 for undiluted sample (row A), 0.1 or 10-1 for the first 1:10 dilution (row B), 0.01 or 10-2 for the second 1:10 dilution (row C), and so on, and the constant 100 accounts for the fact that the total volume of the drop is 10 μL, while the final value is expressed in CFU/mL, i.e., CFU/1,000 μL. Use a spreadsheet containing formulas that calculate CFU/mL from colony count to simplify this process.
- For dilution series where more than one drop was countable (or where two drops had to be counted because no drop fell in range), average the final CFU/mL counts for all counted drops.
- Because the lower limit of detection is 300 CFU/mL (3 colonies in the undiluted drop), record and plot colony count as £300 CFU/mL for dilution series in which there are <3 colonies in the undiluted drop.
- Inspect the sterility control drop from time 24; if any growth is observed in this drop, the results of the experiment should not be used.
- Graph and analyze results.
- Plot growth curves from the three antibiotic-containing cultures and the growth control on the same graph. Plot time on the x axis and CFU/mL, using a logarithmic scale, on the y axis.
- Calculate the difference in CFU/mL between the combination tube at time 24 and the most active single agent at time 24. If the difference is ≥2 log10, consider the combination synergistic. Then calculate the difference in CFU/mL between the combination tube at time 24 and at time 0. If the difference is ≥3 log10, consider the combination bactericidal.
Representative Results
Figure 1A presents a grid from a checkerboard array synergy experiment in which minocycline in concentrations of 0-32 μg/mL was combined with colistin at concentrations of 0-16 μg/mL and tested against E. coli strain FDA-CDC 0494. The values represent spectrophotometric readings at optical density 600 nm (OD600). Wells with OD600 values below 0.07 (which corresponds to no growth by visual inspection) are shaded red, while wells with OD600 values 0.07 (which corresponds to growth by visual inspection) are shaded green. For each drug, the minimum inhibitory concentration (MIC; bolded) is the lowest concentration of drug that inhibits bacterial growth. For minocycline, this is 32 μg/mL, and for colistin, it is 8 μg/mL. The shading is retained in Figure 1B, but values within the wells in which growth is inhibited are replaced by fractional inhibitory concentration index (FICI) values. These are determined as follows: in each well, the fractional inhibitory concentration index (FIC) of each drug is calculated by dividing the concentration of antibiotic in that well by the drug's MIC, and the FICI is calculated by summing the two FICs. Wells with an FICI value of 0.5, which is considered the cutoff for synergy, are indicated with a broken-line border, and the well with the lowest FICI value (0.094) is bolded. Because the minimum FICI value is in the synergistic range, the combination is considered synergistic.
Figure 2A and Figure 2B show grids analogous to those in Figure 1A and Figure 1B, but in this case the combination does not demonstrate synergy against the isolate tested (K. pneumoniae isolate BIDMC 4), because the minimum FICI at which growth is inhibited is 1, which is >0.5.
Figure 3 illustrates the optical density readings from a checkerboard synergy grid in which several skipped wells occurred (Enterobacter cloacae complex isolate BIDMC 27). Skipped wells are wells in which bacterial growth is inhibited despite the presence of bacterial growth in adjacent wells with higher concentrations of antibiotic. This phenomenon, which is known to occur in standard MIC testing as well, is likely due to biological variability in bacterial growth characteristics from well to well and to the sensitivity of some antibiotics to small differences in bacterial inoculum23,31,32. If more than one skipped well occurred in a checkerboard array, we discarded the results and repeated the assay.
Figure 4 presents examples of time-kill synergy results of three combinations tested against K. pneumoniae isolate BIDMC 32. Colony counts are indicated in a logarithmic scale on the y-axis and time, in hours, on the x-axis. The difference between the starting inoculum in the tube containing the drug combination and the concentration of bacteria in that tube at 24 h is illustrated by the red bar and number, while the difference between the concentration of bacteria at 24 h between the tube containing the combination and the tube containing the most active single agent alone is illustrated by the blue bar and number. Figure 4A shows results from the combination of colistin and minocycline; this combination was synergistic (difference between concentrations of bacteria exposed to combination and to most active agent alone ≥2 log10 CFU/mL at 24 h) and bactericidal (decline from starting inoculum to concentration at 24 h ≥3 log10 CFU/mL). Figure 4B shows results from the combination of colistin and clindamycin, a combination that was synergistic but was not bactericidal. This combination inhibited growth of the bacteria, which neither drug did alone, but did not kill them. Figure 4C shows results from the combination of colistin and erythromycin, which was neither bactericidal nor synergistic.
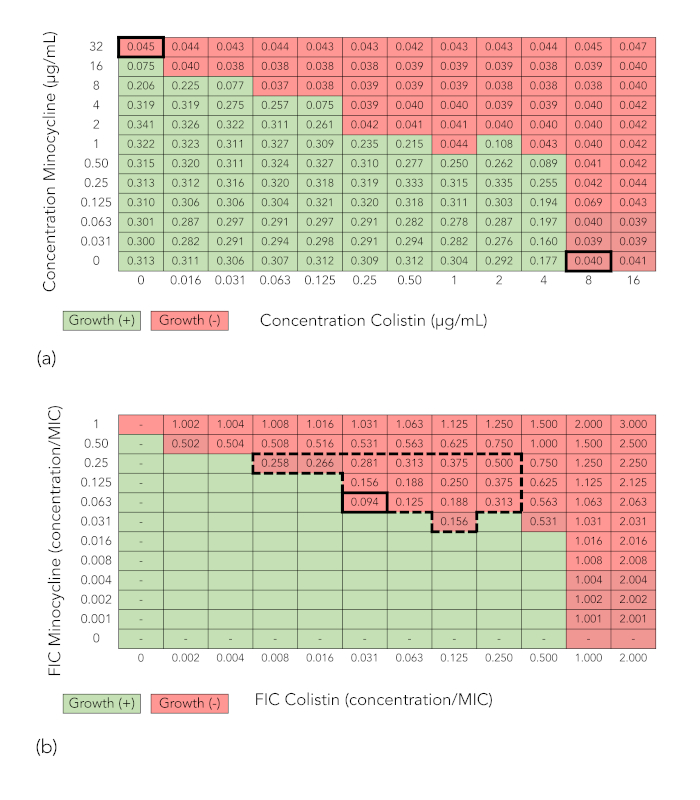
Figure 1: Checkerboard array results demonstrating synergy (minocycline + colistin tested against E. coli strain FDA-CDC 0494). (A) Spectrophotometric readout and growth interpretation of a checkerboard array. Values in cells are optical density readings at 600 nm (OD600). Cells with OD600 values below 0.07 (corresponding to no growth by visual inspection) are shaded red, while cells with OD600 values 0.07 (corresponding to growth by visual inspection) are shaded green. (B) Fractional inhibitory concentration index (FICI) calculation. Shading indicating growth or no growth has been retained. Values for colistin and minocycline along x– and y-axes, respectively, now represent the fractional inhibitory concentration (FIC), or the ratio of the concentration of the drug in that column or row to the minimum inhibitory concentration (MIC) of that drug alone. The value in each cell is the FICI, or the sum of the FICs of the two drugs in that well. The large broken line-bordered box encloses wells with an FICI of 0.5. The thick-bordered cell indicates the well with the lowest FICI in which growth is inhibited, or the minimum FICI. Because the minimum FICI is 0.5, the combination is considered synergistic. Please click here to view a larger version of this figure.
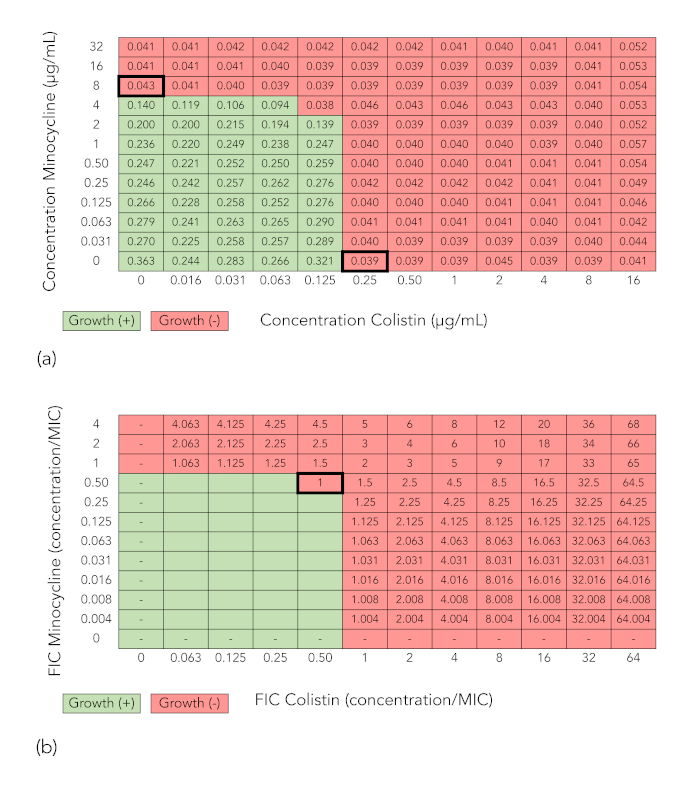
Figure 2: Checkerboard array results of a combination that does not demonstrate synergy (minocycline + colistin tested against K. pneumoniae isolate BIDMC 4). (A) Optical density values at 600 nm and growth interpretation of checkerboard array results as described for Figure 1A. (B) Fractional inhibitory concentration index (FICI) calculation as described for Figure 1A. Because the minimum FICI is >0.5, the combination is not considered synergistic. Please click here to view a larger version of this figure.
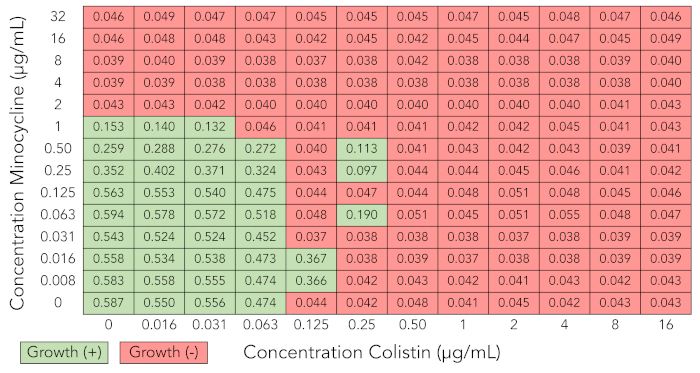
Figure 3: Checkerboard array results that are uninterpretable due to skipped wells (minocycline + colistin tested against Enterobacter cloacae complex isolate BIDMC 27). Optical density values at 600 nm and growth interpretation of checkerboard array results as described for Figure 1A. Several skipped wells, in which bacterial growth is inhibited despite the presence of growth in adjacent wells with higher concentrations of antibiotic, are demonstrated. Results are not interpretable, and experiment needs to be repeated. Please click here to view a larger version of this figure.
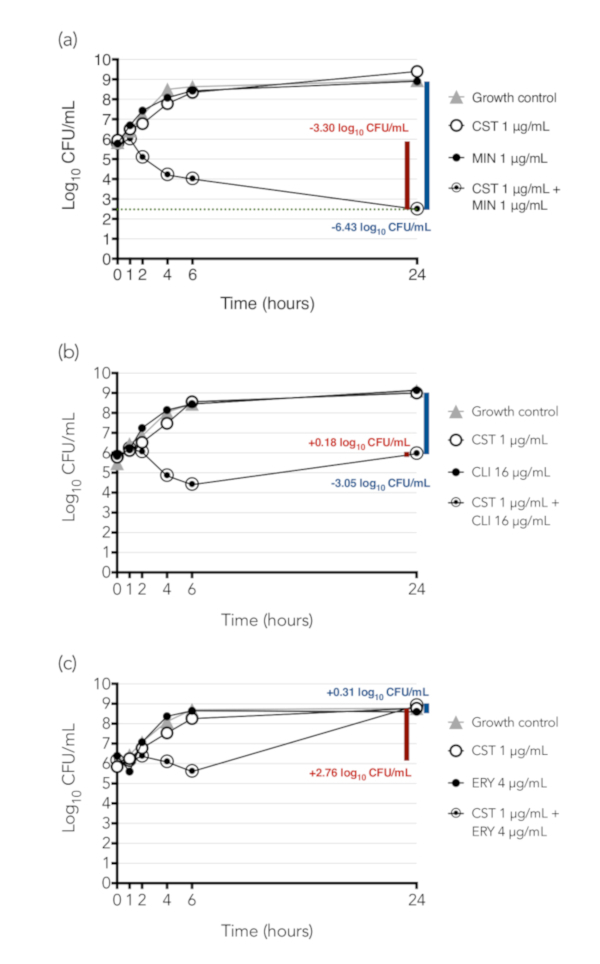
Figure 4: Time-kill synergy results of three combinations tested against K. pneumoniae isolate BIDMC 32. Colony counts are indicated in a logarithmic scale on the y-axis and time, in hours, on the x-axis. The difference between the concentration of bacteria in the combination at 24 h and the starting inoculum in the tube is illustrated by the red bar and number. If the decline from starting inoculum to concentration at 24 h is ≥3 log10 CFU/mL, the combination is considered bactericidal. The difference between the concentration of bacteria at 24 h between the tube containing the combination and the tube containing the most active single agent alone is illustrated by the blue bar and number; if there is ≥2 log10 CFU/mL reduction, the combination is considered synergistic. (A) Colistin (CST) + minocycline (MIN), a combination that is both synergistic and bactericidal. (B) Colistin + clindamycin (CLI), a combination that is synergistic but not bactericidal. (C) Colistin + erythromycin (ERY), a combination that is neither synergistic nor bactericidal. These results were initially published as part of a study of the synergistic activity of colistin-containing combinations against colistin-resistant Enterobacteriaceae, in which we demonstrated that colistin was synergistic with a number of antibiotics that are active individually only (e.g. clindamycin) or primarily (e.g. erythromycin) against Gram-positive bacteria16. (Note that erythromycin was synergistic by checkerboard array against the strain shown, but not by time-kill, so it has been selected here as an example of a non-synergistic combination.) We hypothesized that colistin, which is known to act by permeabilization of the Gram-negative outer membrane, exerts a sub-inhibitory permeabilizing effect on colistin-resistant Gram-negative bacteria, allowing entry of drugs such as clindamycin that normally cannot enter the Gram-negative cell. Panel (A) of this figure has been modified from Brennan-Krohn, Pironti, and Kirby 201816, copyright © American Society for Microbiology, Antimicrobial Agents and Chemotherapy, 62(10), 2018, pii: e00873-18, doi: 10.1128/AAC.00873-18. Please click here to view a larger version of this figure.
Discussion
The two methods described here both provide information about the activity of antimicrobials used in combination compared to their individual activity. The automated, inkjet printer-assisted digital dispensing method is an adaption of the method described in the Clinical Microbiology Procedures Handbook33, while the time-kill method more closely follows the corresponding protocol from the same reference34.
In the checkerboard array method, calculations to determine the necessary volume of antimicrobial stock to add to each well as well as the dispensing of these volumes is automated, thus eliminating some of the major potential sources of error encountered in a manual checkerboard array. It is still essential, however, that the investigator determines that original stocks are made at the intended concentration and that goal final concentrations are entered into the D300 software correctly. Adding the antimicrobial suspension to wells in a 384-well plate can be challenging at first and requires care to ensure that pipette tips enter the appropriate wells and that liquid does not splash up the edges of the wells. An automated liquid handler can be used in place of a hand-held multichannel pipette to increase the speed and accuracy with which the bacterial suspension is added to wells. As described in the protocol, the D300 requires the addition of the surfactant, polysorbate 20 (P-20), for proper liquid handling. A different surfactant, polysorbate 80, at a concentration of 0.002%, has been noted to lower colistin MICs for organisms with colistin MICs of <2 µg/mL in standard broth microdilution assays.35,36 Our laboratory previously demonstrated that P-20 at concentrations up to 0.0015% had no effect on D300-assisted MIC results in comparison with reference BMD14. In the assay example presented here, the maximum P-20 is concentration is 0.0014%.
One problem we encountered with some checkerboard array assays was a large number of skipped wells. This occurred at a disproportionate rate with certain antibiotics. Specifically, in a screen of combinations against a collection of carbapenem-resistant Enterobacteriaceae, we found that while 49 of 521 trials (9.4%) were unusable due to multiple skipped wells, 2 of the 12 antibiotics tested (fosfomycin and cefepime) accounted for 46 of these trials (94%). Such increased rates may be more likely in drugs that are particularly susceptible to the inoculum effect31,32,37. Of note, CLSI does not recommend testing fosfomycin in broth dilution25 due to concerns about the reliability of results with this method, which may explain the unreliable results seen with this drug. Some modifications can be made to automated checkerboard method according to investigator preference. Antimicrobials can be dispensed into plates already containing bacterial suspension, rather than into empty wells, if this is preferable for reasons of workflow within the laboratory. While 384-well plates were used here, the method can also be carried out in 96-well plate assays with appropriate modification of well volume. The use of a 96-well plate format may help in reducing skipped wells for antibiotics that are particularly sensitive to small changes in inoculum. When calculating FICI, there may be situations where the MIC is off-scale (i.e., higher than tested), including situations where the drug being tested has no activity individually against the type of organism being tested. In these cases, the FIC can be calculated based on assuming the MIC is one dilution higher than the highest concentration tested. This is the most conservative strategy, as it assumes the maximal possible FIC value for any dilution where inhibition is observed during synergy testing. For example, if the actual MIC were instead two doubling dilutions above the highest concentration tested, then the corresponding FICs would be two-fold lower than the conservative assignments, and so on.
In order to accurately assess the bactericidal activity of drugs in a time-kill assay, it is essential that cultures be in logarithmic-phase growth, particularly when cell-wall active antibiotics are being tested28. For the rapidly-growing bacteria used in this example (K. pneumoniae), 3 hours of incubation with shaking was appropriate to reach this growth phase, but different amounts of time may be necessary for different organisms. In general, the culture should appear visibly but not heavily turbid. The appropriate amount of time can be determined by constructing a growth curve with colony counts taken at serial time points (e.g., every 30 min for 4-6 h)38. The intended starting inoculum in the time-kill study is also important. The target concentration of the starting inoculum is approximately 5 x 105 to 1 x 106 CFU/mL. The dilution described here (100 μL of a 1.0 McFarland suspension in 10 mL of media) generates this inoculum for Klebsiella pneumoniae and other Enterobacteriaceae species on which we have tested it. If the density of the starting inocula in an experiment using different organisms is significantly higher or lower than this, then a different dilution may be needed. (The appropriate dilution required for a given species can be determined by performing a plate count of a 0.5 or 1.0 McFarland suspension to determine how many organisms this turbidity represents, then calculating the amount by which the initial suspension must be diluted to reach the appropriate final concentration.) If, on review of plate counts from the synergy study, the starting inoculum of any of the antibiotic-containing tubes is found to have been significantly lower than the starting inoculum of the growth control, this may indicate either antibiotic carryover or very rapid killing of bacteria in the brief time between addition of bacteria to the antibiotic-containing tube and removal of the aliquot for plating. If the actual number of colonies in the undiluted drop in a series is lower than the number of colonies in subsequent dilutions, this suggests antibiotic carryover effect. Different options have been described for preventing this effect, including spreading a single aliquot over an entire plate38 or spinning down the sample, removing the supernatant, and re-suspending in sterile saline prior to plating39. At each time point in the time-kill method, it is also critical for the investigator to efficiently but accurately remove an aliquot from each culture tube and perform serial dilutions. Delays during this process, particularly during early time points that occur in close succession, can lead to prolonged periods during which cultures are not been incubated and shaken, whereas careless dispensing and serial dilutions can lead to inaccurate plate counts. Compared to the spread plate method of plate counting, in which 100 μL of each dilution is spread over an entire agar plate, the drop plate method described is far more rapid, requires a much smaller number of agar plates, and allows for faster counting, as the maximum countable number of colonies for each drop is 30, whereas up to 300 colonies can typically be counted from a spread plate. However, the spread plate method is also an option if investigators are more comfortable with this technique. If drops spread into each other after dispensing with a multichannel pipette, individual application of more widely spaced drops with a single-channel pipette can be performed instead. In our experience, cooling plates at 4 °C prior to dispensing drops seemed to reduce excessive spreading.
One limitation of the techniques described here is that the results of the two types of synergy assay (checkerboard array and time-kill) are not always concordant, and since most published synergy articles use one method or the other rather than both together, it can be difficult to know how to integrate data from the two types of assays. Because the automated checkerboard array method we developed is simple and high-throughput, we have used it in effect as a kind of screen to test combinations against a larger number of isolates and to determine which concentration combinations were synergistic. We then performed a smaller number of time-kill studies, selecting combinations and concentrations that had been effective in the checkerboard array. Of note, because the checkerboard assay is typically performed on a microbroth dilution scale, while the time-kill assay uses larger volumes (similar to a macrobroth dilution), we found that FICs were sometimes different between the two methods, with higher concentrations generally required in the time-kill assay to demonstrate activity. This phenomenon has been noted previously when macrobroth and microbroth dilution MIC assay results are compared for Gram-negative bacilli26 and when larger inocula (as used in time-kill studies) are compared with the standard inoculum used in microbroth dilution and checkerboard array assays32. A specific limitation of the checkerboard array is the inherent variability in microbroth dilution MIC testing22. While FICI cutoffs for synergy account for this variability mathematically6 such variability inevitably raises concern about the reliability and consistency of checkerboard array results.
Because of the limitations inherent to all in vitro synergy testing methods (including cultivation of bacteria in an artificial growth medium, static antibiotic concentrations, and a limited time course), results obtained by these methods must be confirmed and further evaluated using supplemental techniques. Such methods include in vitro pharmacokinetic/pharmacodynamic (PK/PD) studies (e.g., the hollow fiber infection model40), animal models, and, ultimately, human PK/PD and efficacy studies. The automated checkerboard array method described here, by providing a rapid method with which to screen combinations for potential synergistic activity, allows for more targeted utilization of these techniques. Further automation of all of these methods, as well as more systematic investigation of the relationship between in vitro parameters and clinical outcomes, will be important in scaling up the use of synergy testing and increasing its clinical applicability.
Disclosures
The authors have nothing to disclose.
Acknowledgements
Thea Brennan-Krohn was supported by a Eunice Kennedy Shriver National Institute of Child Health and Human Development pediatric infectious diseases research training grant (T32HD055148), a National Institute of Allergy and Infectious Diseases training grant (T32AI007061), a Boston Children's Hospital Office of Faculty Development Faculty Career Development fellowship, and a National Institute of Allergy and Infectious Diseases career development award (1K08AI132716). J.E.K. was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number R33 AI119114. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Materials
| Escherichia coli strain ATCC 25922 | ATCC | 25922 | QC strain |
| 0.5 mL microcentrifuge tubes | USA Scientific | 1605-0000 | |
| 1 L 0.22 µm bottle-top filter | Thermo Scientific Nalgene | 597-4520 | |
| 12 mm x 75 mm borosilicate glass round bottom culture tubes | Fisherbrand | 14-961-26 | |
| 15 mL conical tubes | Phenix | SS-PH15 | |
| 15 x 100 mm or 15 x 150 mm Mueller Hinton agar plates | Thermo Scientific | R01620 or R04050 | |
| 25 mm stainless steel closures for 25 x 150 mm glass culture tubes | Bellco | 2005-02512 | |
| 25 x 150 mm borosilicate glass round bottom culture tubes | Bellco | 2011-25150 | |
| 348-well sterile clear, flat-bottom, untreated microplates with lids | Greiner Bio-One | 781186 | |
| 50 mL conical tubes | Phenix | SS-PH50 | |
| 50 mL sterile reagent reservoirs | Corning | 4870 | |
| 96 deep well polypropylene microplate with 2 mL wells | Fisherbrand | 12-566-612 | |
| 96-well sterile clear, round-bottom, untreated microplates with lids | Evergreen | 222-8032-01R | |
| Cation adjusted Mueller Hinton broth | BD Diagnostics | 212322 | |
| Colistin sulfate | Alfa Aesar | J60915 | |
| D300e Control Software | HP/Tecan | ||
| DensiCHEK Plus McFarland reader | bioMérieux | 21250 | |
| Excel spreadsheet software | Microsoft | ||
| Extra long SHARP 10 µL Precision Barrier Tips | Denville Scientific | P1096-FR | |
| HP D300 digital dispenser | HP/Tecan | ||
| HP D300 T8+ cassettes | HP/Tecan | 30097370 | |
| Minocycline hydrochloride | Chem-Impex | 14302 | |
| Picus 12-channel 10-300 µL pipette | Sartorius | 735461 | |
| Polysorbate 20 | Fisher Bioreagents | BP-337 | Brand name: Tween 20 |
| Sodium chloride | Fisher Chemical | S271 | |
| Spectrophotometer | Tecan | Infinite M1000 PRO | |
| Xplorer 12-channel 50-1200 µL pipette | Eppendorf | 2231000328 |
References
- Temkin, E., Adler, A., Lerner, A., Carmeli, Y. Carbapenem-resistant Enterobacteriaceae: Biology, epidemiology, and management. Annals of the New York Academy of Sciences. 1323 (1), 22-42 (2014).
- Spellberg, B. The future of antibiotics. Critical Care. 18 (3), (2014).
- Spellberg, B., et al. The epidemic of antibiotic-resistant infections: a call to action for the medical community from the Infectious Diseases Society of America. Clinical infectious diseases an official publication of the Infectious Diseases Society of America. 46 (2), 155-164 (2008).
- Elemam, A., Rahimian, J., Doymaz, M. In vitro evaluation of antibiotic synergy for polymyxin B-resistant carbapenemase-producing Klebsiella pneumoniae. Journal of Clinical Microbiology. 48 (10), 3558-3562 (2010).
- Poirel, L., Kieffer, N., Nordmann, P. In vitro evaluation of dual carbapenem combinations against carbapenemase-producing Enterobacteriaceae. Journal of Antimicrobial Chemotherapy. 71 (1), 156-161 (2016).
- Odds, F. C. Synergy, antagonism, and what the chequerboard puts between them. Journal of Antimicrobial Chemotherapy. 52, (2003).
- Zusman, O., et al. Systematic review and meta-analysis of in vitro synergy of polymyxins and carbapenems. Antimicrobial Agents and Chemotherapy. 57 (10), 5104-5111 (2013).
- Clock, S. A., et al. In vitro activity of doripenem alone and in multi-agent combinations against extensively drug-resistant Acinetobacter baumannii and Klebsiella pneumoniae. Diagnostic Microbiology and Infectious Disease. 76 (3), 343-346 (2013).
- Hirsch, E. B., et al. Assessment of antimicrobial combinations for Klebsiella pneumoniae carbapenemase-producing K. pneumoniae. Journal of Infectious Diseases. 207 (5), 786-793 (2013).
- Tängdén, T., et al. Evaluation of double- and triple-antibiotic combinations for VIM- and NDM-producing klebsiella pneumoniae by in vitro time-kill experiments. Antimicrobial Agents and Chemotherapy. 58 (3), 1757-1762 (2014).
- Tascini, C., et al. Synergistic activity of colistin plus rifampin against colistin-resistant kpc-producing klebsiella pneumoniae. Antimicrobial Agents and Chemotherapy. 57 (8), 3990-3993 (2013).
- Paul, M., et al. Combination therapy for carbapenem-resistant Gram-negative bacteria. Journal of Antimicrobial Chemotherapy. 69 (9), 2305-2309 (2014).
- Rafailidis, P. I., Falagas, M. E. Options for treating carbapenem-resistant Enterobacteriaceae. Current Opinion in Infectious Diseases. 27 (6), 479-483 (2014).
- Smith, K. P., Kirby, J. E. Verification of an automated, digital dispensing platform for at-will broth microdilution-based antimicrobial susceptibility testing. Journal of Clinical Microbiology. 54 (9), 2288-2293 (2016).
- Brennan-Krohn, T., Truelson, K., Smith, K. P., Kirby, J. E. Screening for Synergistic Activity of Antimicrobial Combinations Against Carbapenem-Resistant Enterobacteriaceae Using Inkjet Printer-Based Technology. J Antimicrob Chemother. 72 (10), 2775-2781 (2017).
- Brennan-Krohn, T., Pironti, A., Kirby, J. E. Synergistic Activity of Colistin-Containing Combinations against Colistin-Resistant Enterobacteriaceae. Antimicrobial Agents and Chemotherapy. , (2018).
- Falagas, M. E., Kasiakou, S. K., Saravolatz, L. D. Colistin: The Revival of Polymyxins for the Management of Multidrug-Resistant Gram-Negative Bacterial Infections. Clinical Infectious Diseases. 40 (9), 1333-1341 (2005).
- Nation, R. L., Li, J. Colistin in the 21st century. Current Opinion in Infectious Diseases. 22 (6), 535-543 (2009).
- Ah, Y. -. M., Kim, A. -. J., Lee, J. -. Y. Colistin resistance in Klebsiella pneumoniae. International Journal of Antimicrobial Agents. 44 (1), 8-15 (2014).
- Bratu, S., et al. Carbapenemase-producing Klebsiella pneumoniae in Brooklyn, NY: Molecular epidemiology and in vitro activity of polymyxin B and other agents. Journal of Antimicrobial Chemotherapy. 56 (1), 128-132 (2005).
- Barth, N., Ribeiro, V. B., Zavasckid, A. P. In vitro activity of polymyxin B plus imipenem, meropenem, or tigecycline against KPC-2-producing Enterobacteriaceae with high MICs for these antimicrobials. Antimicrobial Agents and Chemotherapy. 59 (6), 3596-3597 (2015).
- MacLowry, J., Jaqua, M., Selepak, S. Detailed methodology and implementation of a semiautomated serial dilution microtechnique for antimicrobial susceptibility testing. Appl Microbiol. 20 (1), 46-53 (1970).
- Brennan-Krohn, T., Smith, K. P., Kirby, J. E. The poisoned well: Enhancing the predictive value of antimicrobial susceptibility testing in the era of multidrug resistance. Journal of Clinical Microbiology. 55 (8), 2304-2308 (2017).
- Doern, C. D. When does 2 plus 2 equal 5? A review of antimicrobial synergy testing. Journal of Clinical Microbiology. 52 (12), 4124-4128 (2014).
- CLSI. . Performance Standards for Antimicrobial Susceptibility Testing. 28th ed. CLSI supplement M100. , (2018).
- CLSI. . Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard – Tenth Edition. CLSI document M07-A10. , (2015).
- Clinical and Laboratory Standards Institute. . M07: Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically, 11th Edition. , (2018).
- Clinical and Laboratory Standards Institute. . Methods for determining bactericidal activity of antimicrobial agents; approved guideline M26-A. 19 (18), 7 (1999).
- Naghili, H., Tajik, H., Mardani, K., Razavi Rouhani, S. M., Ehsani, A., Zare, P. Validation of drop plate technique for bacterial enumeration by parametric and nonparametric tests. Veterinary research forum. 4 (3), 179-183 (2013).
- Chen, C. Y., Nace, G. W., Irwin, P. L. A 6×6 drop plate method for simultaneous colony counting and MPN enumeration of Campylobacter jejuni, Listeria monocytogenes, and Escherichia coli. Journal of Microbiological Methods. 55 (2), 475-479 (2003).
- Queenan, A. M., Foleno, B., Gownley, C., Wira, E., Bush, K. Effects of Inoculum and Activity in AmpC- and Extended-Spectrum (ESBL)-Producing Escherichia coli and Klebsiella pneumoniae Clinical Isolates Tested by Using NCCLS ESBL Methodology. Journal of Clinical Microbiology. 42 (1), 269-275 (2004).
- Smith, K. P., Kirby, J. E. The Inoculum Effect in the Era of Multidrug Resistance: Minor Differences in Inoculum Have Dramatic Effect on Minimal Inhibitory Concentration Determination. Antimicrobial Agents and Chemotherapy. , (2018).
- Leber, A. L. Synergism Testing: Broth Microdilution Checkerboard and Broth Macrodilution Methods. Clinical Microbiology Procedures Handbook, Fourth Edition. , (2016).
- Leber, A. L. Time-Kill Assay for Determining Synergy. Clinical Microbiology Procedures Handbook, Fourth Edition. , (2016).
- Hindler, J. A., Humphries, R. M. Colistin MIC variability by method for contemporary clinical isolates of multidrug-resistant gram-negative bacilli. Journal of Clinical Microbiology. 51 (6), 1678-1684 (2013).
- Sutherland, C. A., Nicolau, D. P. To add or not to add Polysorbate 80: Impact on colistin MICs for clinical strains of Enterobacteriaceae and Pseudomonas aeruginosa and quality controls. Journal of Clinical Microbiology. 52 (10), 3810 (2014).
- Fuchs, P. C., Barry, a. L., Brown, S. D. Susceptibility testing quality control studies with fosfomycin tromethamine. European journal of clinical microbiology & infectious diseases official publication of the European Society of Clinical Microbiology. 16 (7), 538-540 (1997).
- Leber, A. L. Minimum Bactericidal Concentration Testing. Clinical Microbiology Procedures Handbook, Fourth Edition. 5.14.1.11. , (2016).
- Cai, Y., et al. In vitro activity of polymyxin B in combination with various antibiotics against extensively drug-resistant Enterobacter cloacae with decreased susceptibility to polymyxin B. Antimicrobial Agents and Chemotherapy. 60 (9), 5238-5246 (2016).
- Bulman, Z. P., et al. Polymyxin combinations combat Escherichia coli harboring mcr-1 and blaNDM-5: Preparation for a postantibiotic Era. mBio. 8 (4), (2017).
