En las últimas dos décadas, sólidos y películas que están hechas de moléculas orgánicas agregadas han encontrado múltiples aplicaciones en dispositivos optoelectrónicos. Los dispositivos basados en estos materiales tienen muchas propiedades atractivas, incluyendo peso pequeño, flexibilidad, bajo consumo de energía y potencial de producción barata utilizando la impresión de inyección de tinta. Las pantallas basadas en diodos orgánicos emisores de luz (OLED) están reemplazando las pantallas cristalinas líquidas como estado de la técnica para teléfonos móviles, portátiles, televisores y otros dispositivos electrónicos1,2,3,4. Se espera que la importancia de los ODOpara las aplicaciones de iluminación aumente en los próximos años4. El rendimiento de los dispositivos fotovoltaicos orgánicos está mejorando constantemente, con eficiencias de conversión de energía por encima del 16% recientemente reportadas para células solares orgánicas de unión única5. Los materiales orgánicos también tienen el potencial de interrumpir otras tecnologías, como las comunicaciones de fibra óptica, donde su uso permite el desarrollo de moduladores electroópticos con anchos de banda extremadamente altos de 15 THz y superioresa6,7.
Un desafío importante en la optimización de materiales moleculares de estado sólido para aplicaciones en optoelectrónica es que normalmente sus propiedades dependen en gran medida de la estructura a nanoescala del material. El proceso de producción permite definir la nanoestructura de un material en cierta medida mediante el uso de técnicas de crecimiento controlado, tales como la deposición de vapor químico,8 plantillas de moléculas ópticamente activas en otro material (es decir, una matriz depolímeros 9,10), recocido térmico11,,12,etc. Sin embargo, el trastorno a nanoescala es intrínseco a la mayoría de los materiales moleculares y por lo general no se puede eliminar por completo. Por lo tanto, entender cómo el trastorno afecta las propiedades de un material y encontrar maneras de diseñarlo para un rendimiento óptimo es esencial para el diseño racional de materiales optoelectrónicos orgánicos.
El grado de trastorno en los materiales moleculares suele ser demasiado grande para tratarlo como una perturbación de una estructura cristalina periódica con una estructura electrónica que puede ser descrita por la teoría de la banda. Por otro lado, el número de moléculas que deben incluirse en una simulación para reproducir las propiedades de un material a granel o una película es demasiado grande para utilizar los primeros principios métodos químicos cuánticos como la teoría funcional de densidad (DFT)13,14 y la teoría funcional de densidad dependiente del tiempo (TD-DFT)15,16. Las moléculas orgánicas con aplicaciones en optoelectrónica suelen tener sistemas relativamente grandes conjugados; muchos también tienen grupos de donantes y aceptadores. Capturar el comportamiento correcto de transferencia de carga en estas moléculas es esencial para calcular sus propiedades optoelectrónicas, pero sólo se puede lograr utilizando funciones híbridas corregidas de largo alcance en TD-DFT17,,18,,19,,20. Los cálculos que utilizan tales funciones se escalan súper linealmente con el tamaño del sistema y, en la actualidad, sólo son prácticos para modelar las propiedades optoelectrónicas de moléculas orgánicas individuales o pequeños agregados moleculares que se pueden describir utilizando no más de 104 funciones de base atómica. Un método de simulación que podría describir materiales desordenados que consisten en un gran número de cromóforos sería muy útil para modelar estos sistemas.
La magnitud de las interacciones intermoleculares en los materiales moleculares es a menudo comparable o menor que el orden de variación en los parámetros energéticos (como las energías eigenestatales o las energías de excitación) entre moléculas individuales que componen el material. En tales casos, el modelado multiescala es el enfoque más prometedor para comprender y optimizar las propiedades optoelectrónicas de los grandes sistemas moleculares desordenados21,,22,,23. Este enfoque utiliza métodos químicos cuánticos de primeros principios (normalmente DFT y TD-DFT) para calcular con precisión las propiedades de las moléculas individuales que componen el material. El Hamiltoniano de una muestra de material que es lo suficientemente grande como para representar el material molecular a granel (tal vez, mediante el empleo de condiciones límite periódicas) se construye utilizando los parámetros que se calcularon para moléculas individuales. Este Hamiltoniano se puede utilizar para calcular los parámetros optoelectrónicos de un agregado molecular grande, una película delgada o un material molecular a granel.
Los modelos exciton son una subclase de modelos multiescala en los que los estados excitados de un material molecular se representan en base a exciminutos:pares de electrones-agujeros que están unidos por la atracción Coulomb24,25. Para modelar muchos procesos de estado excitado, es suficiente incluir sólo excitons de Frenkel26,donde el electrón y el agujero se localizan en la misma molécula. Los excicranos de transferencia de carga, donde el electrón y el agujero están localizados en diferentes moléculas, pueden necesitar ser incluidos en algunos casos (por ejemplo, al modelar la separación de carga en sistemas de donante-aceptador)27,28. Aunque los modelos de excitón son modelos multiescala que se pueden parametrizar utilizando sólo cálculos de primer principio en moléculas individuales, todavía tienen en cuenta las interacciones intermoleculares. Los dos tipos de interacción principales que pueden explicar son (a) acoplamientos excitónicos entre moléculas que caracterizan la capacidad de los excicidores para deslocalizar o transferir entre moléculas y (b) polarización electrostática de la densidad de electrones en una molécula por la distribución de carga en las moléculas circundantes. Hemos demostrado previamente que ambos factores son importantes para modelar las propiedades ópticas y electroópticas de los agregados moleculares, como los espectros de absorción óptica29 y las primeras hiperpolarizabilidades30.
En este artículo, presentamos un protocolo para parametrizar modelos de exciton que se pueden utilizar para calcular los espectros ópticos y otras propiedades optoelectrónicas de grandes agregados moleculares y materiales moleculares a granel. Se supone que el Hamiltoniano excitónico es un Hamiltoniano de24,,25,
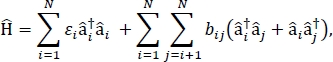
donde i es la energía de excitación de la molécula ith en el material, bij es el acoplamiento excitónico entre las moléculas ith y jth, i y i son los operadores de creación y aniquilación, respectivamente, para un estado excitado en la molécula ith en el material. i† Los parámetros hamiltonianos excitónicos se encuentran utilizando cálculos TD-DFT que se realizan en moléculas individuales que componen el material. En estos cálculos TD-DFT, la distribución de la carga en todas las demás moléculas del material está representada por la incrustación electrostática de cargas de punto atómico para tener en cuenta la polarización electrostática de la densidad electrónica de una molécula. Las energías de excitación, i, para moléculas individuales se toman directamente de la salida de cálculo TD-DFT. Los acoplamientos excónicos, bij, entre moléculas se calculan utilizando el método de cubo de densidad de transición31,con las densidades de transición de estado de tierra a excitación para las moléculas que interactúan tomadas de la salida de un cálculo TD-DFT en Gaussian32 y posteriores al procesamiento utilizando el analizador multifunción de onda33. Multiwfn Para simular las propiedades de los sólidos moleculares a granel, se pueden aplicar condiciones límite periódicas al Hamiltoniano.
El protocolo actual requiere que el usuario tenga acceso a los programas Gaussian32 y Multiwfn33. El protocolo ha sido probado usando Gaussian 16, revisión B1 y Multiwfn versión 3.3.8, pero también debería funcionar para otras versiones recientes de estos programas. Además, el protocolo utiliza una utilidad C++ personalizada y una serie de scripts personalizados de Python 2.7 y Bash, cuyo código fuente se proporciona bajo la Licencia Pública General GNU (Versión 3) en https://github.com/kocherzhenko/ExcitonicHamiltonian. Los cálculos están diseñados para realizarse en una máquina que ejecuta un sistema operativo de la familia Unix/Linux.
