In the past two decades, solids and films that are made from aggregated organic molecules have found multiple applications in optoelectronic devices. Devices based on such materials have many attractive properties, including small weight, flexibility, low power consumption, and potential for cheap production using inkjet printing. Displays based on organic light emitting diodes (OLEDs) are replacing liquid crystalline displays as state of the art for mobile phones, laptops, television sets, and other electronic devices1,2,3,4. The importance of OLEDs for lighting applications is expected to increase in the coming years4. The performance of organic photovoltaic devices is steadily improving, with power conversion efficiencies above 16% recently reported for single-junction organic solar cells5. Organic materials also have the potential to disrupt other technologies, such as fiber-optic communications, where their use enables the development of electro-optic modulators with extremely high bandwidths of 15 THz and above6,7.
A major challenge in optimizing solid-state molecular materials for applications in optoelectronics is that typically their properties strongly depend on the nanoscale structure of the material. The production process allows defining the nanostructure of a material to some extent by using controlled growth techniques, such as chemical vapor deposition,8 templating of optically active molecules onto another material (i.e., a polymer matrix9,10), thermal annealing11,12, etc. However, nanoscale disorder is intrinsic to most molecular materials and usually cannot be eliminated entirely. Therefore, understanding how disorder affects the properties of a material and finding ways to engineer it for optimal performance is essential for the rational design of organic optoelectronic materials.
The degree of disorder in molecular materials is usually too great to treat it as a perturbation of a periodic crystalline structure with an electronic structure that can be described by band theory. On the other hand, the number of molecules that must be included in a simulation to reproduce the properties of a bulk material or a film is too great to use first principles quantum chemical methods like density functional theory (DFT)13,14 and time-dependent density functional theory (TD-DFT)15,16. Organic molecules with applications in optoelectronics typically have relatively large π-conjugated systems; many also have donor and acceptor groups. Capturing the correct charge-transfer behavior in such molecules is essential to calculating their optoelectronic properties, but it can only be accomplished using long-range corrected hybrid functionals in TD-DFT17,18,19,20. Calculations that use such functionals scale super linearly with the size of the system and, at present, they are only practical for modeling the optoelectronic properties of individual organic molecules or small molecular aggregates that can be described using no more than ~104 atomic basis functions. A simulation method that could describe disordered materials that consist of large numbers of chromophores would be very useful for modeling these systems.
The magnitude of intermolecular interactions in molecular materials is often comparable to or smaller than the order of variation in the energetic parameters (such as the eigenstate energies or excitation energies) between individual molecules that make up the material. In such cases, multiscale modeling is the most promising approach to understanding and optimizing the optoelectronic properties of large disordered molecular systems21,22,23. This approach uses first-principles quantum chemical methods (usually DFT and TD-DFT) to accurately calculate the properties of individual molecules that compose the material. The Hamiltonian of a material sample that is large enough to represent the bulk molecular material (perhaps, by employing periodic boundary conditions) is then constructed using the parameters that were calculated for individual molecules. This Hamiltonian can then be used to calculate the optoelectronic parameters of a large molecular aggregate, a thin film, or a bulk molecular material.
Exciton models are a subclass of multiscale models in which excited states of a molecular material are represented in a basis of excitons: electron-hole pairs that are bound by Coulomb attraction24,25. For modeling many excited state processes, it is sufficient to only include Frenkel excitons26, where the electron and the hole are localized on the same molecule. Charge transfer excitons, where the electron and the hole are localized on different molecules, may need to be included in some cases (e.g., when modeling charge separation in donor-acceptor systems)27,28. Although exciton models are multiscale models that can be parametrized using only first-principle calculations on individual molecules, they still account for intermolecular interactions. The two primary interaction types that they can account for are (a) excitonic couplings between molecules that characterize the ability of excitons to delocalize across or transfer between molecules and (b) electrostatic polarization of the electron density on a molecule by the charge distribution on surrounding molecules. We have previously shown that both of these factors are important for modeling the optical and electro-optic properties of molecular aggregates, such as the optical absorption spectra29 and first hyperpolarizabilities30.
In this paper, we present a protocol for parametrizing exciton models that can be used to calculate the optical spectra and other optoelectronic properties of large molecular aggregates and bulk molecular materials. The excitonic Hamiltonian is assumed to be a tight-binding Hamiltonian24,25,
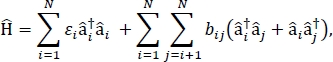
where εi is the excitation energy of the ith molecule in the material, bij is the excitonic coupling between the ith and the jth molecules, âi† and âi are the creation and annihilation operators, respectively, for an excited state on the ith molecule in the material. The excitonic Hamiltonian parameters are found using TD-DFT calculations that are performed on individual molecules that make up the material. In these TD-DFT calculations, the charge distribution on all other molecules in the material is represented by electrostatic embedding of atomic point charges to account for electrostatic polarization of a molecule’s electronic density. The excitation energies, εi, for individual molecules are taken directly from the TD-DFT calculation output. The excitonic couplings, bij, between molecules are calculated using the transition density cube method31, with the ground-to-excited state transition densities for the interacting molecules taken from the output of a TD-DFT calculation in Gaussian32 and post-processed using the Multiwfn multifunctional wavefunction analyzer33. For simulating the properties of bulk molecular solids, periodic boundary conditions may be applied to the Hamiltonian.
The current protocol requires that the user have access to the Gaussian32 and Multiwfn33 programs. The protocol has been tested using Gaussian 16, revision B1 and Multiwfn version 3.3.8, but should also work for other recent versions of these programs. In addition, the protocol uses a custom C++ utility and a number of custom python 2.7 and Bash scripts, the source code for which is provided under the GNU General Public License (Version 3) at https://github.com/kocherzhenko/ExcitonicHamiltonian. The calculations are intended to be performed on a machine running an operating system from the Unix/Linux family.
In this section we present representative results for computing the optical absorption spectrum of an aggregate of six YLD 124 molecules, shown in Figure 3a, where the structure of the aggregate was obtained from a coarse-grained Monte Carlo simulation. YLD 124 is a prototypical charge-transfer chromophore that consists of an electron-donating group of diethyl amine with tert-butyldimethylsilyl protecting groups that is connected via a π -conjugated bridge to the electron accepting group 2-(3-cyano-4,5,5-trimethyl-5H-furan-2-ylidene)-malononitrile39. This molecule has a large ground-state dipole moment, ~30 D. Electronic structure calculations for individual molecules were performed using the ωB97X34 functional with the 6-31G* basis set35,36. TD-DFT calculations used the Tamm-Dancoff approximation40. Partial atomic charges were computed with the CHelpG population analysis method37.
The Hamiltonian for this system, constructed using the protocol described in this paper, is shown in Table 1.
The absorption spectrum calculated for this excitonic Hamiltonian is shown in blue in Figure 3b. Because there are six molecules with only a single bright excited state for each molecule, a 6-by-6 excitonic Hamiltonian was generated, resulting in six transitions. The eigenvalues of this Hamiltonian are the lowest six excited state energies for the molecular aggregate. The height of the vertical lines represents the oscillator strength fi for each transition from the ground to the ith excited state of the molecular aggregate. It can be found using the expression29
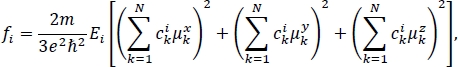
where m is the electron mass, e is the elementary charge, ħ is the reduced Planck’s constant, N is the total number of molecules in the aggregate, Ei is the eigenvalue that corresponds to the ith excited state of the molecular aggregate, cki is the expansion coefficient for the contribution of the kth molecule in the aggregate to the ith excited state of the aggregate written in the basis of bright excited states on individual molecules, and μkα are the components of the transition dipole moment vector for the ground to bright excited state of the kth molecule in the aggregate, α = x, y, z . The values of Ei and cki are found by solving the eigenvalue equation for the Hamiltonian matrix (the time-independent Schrödinger equation). The values of μkα can be found in the “.log2” files that are generated in step 5.2 of the protocol. The total spectrum is a smooth line created by summing over Gaussian functions centered at each of the excitation energies and weighted by the corresponding oscillator strengths29.
For comparison, the spectrum computed from an all-electron TD-DFT calculation on the entire molecular aggregate is shown in magenta. For these spectra, the integrated intensity of the exciton spectrum is larger than the TD-DFT spectrum (Iexc/ITD-DFT = 1.124) and the difference in the mean absorption energies is Eexc – ETD-DFT = 0.094 eV. These offsets are systematic for molecular aggregates of a given size and can be corrected for to obtain very good agreement between exciton model and TD-DFT spectra. For instance, for a set of 25 molecular aggregates that each consists of 6 YLD 124 molecules, the average integrated intensity ratio Iexc/ITD-DFT = 1.126, with a standard deviation of 0.048, and the difference in the mean absorption energies is Eexc – ETD-DFT = 0.057 eV, with a standard deviation of 0.017 eV. The exciton model and TD-DFT spectra shown in Figure 3b also have similar shapes, as characterized by Pearson’s product-moment correlation coefficient41 between them of 0.9818 and Pearson’s product-moment correlation coefficient between their derivatives of 0.9315. On average, for a set of 25 molecular aggregates that each consists of 6 YLD 124 molecules, the agreement in spectral shape is even better than for the example shown, with values of 0.9919 (standard deviation of 0.0090) and 0.9577 (standard deviation of 0.0448) for the two Pearson’s coefficients, respectively29. Our earlier work suggests that the spectral shape is primarily determined by local electrostatic interactions between chromophores in the aggregate that are accounted for in the exciton model described in this paper, whereas the excitation energy and intensity depend considerably on the mutual polarization between the chromophore and its environment that the model neglects29.

Figure 1: An isosurface for the transition density plotted for a single molecule of YLD 124. The the positions of atomic charges on surrounding molecules are shown by gray dots. Please click here to view a larger version of this figure.
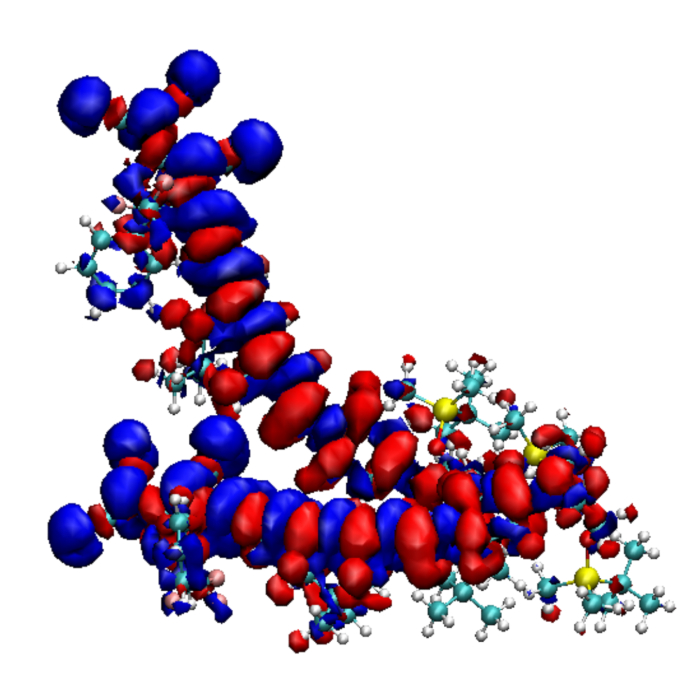
Figure 2: The transition densities plotted for two molecules of YLD 124, i and j, that are used for computing the excitonic coupling bij between these molecules. The surrounding charges are not shown. Please click here to view a larger version of this figure.
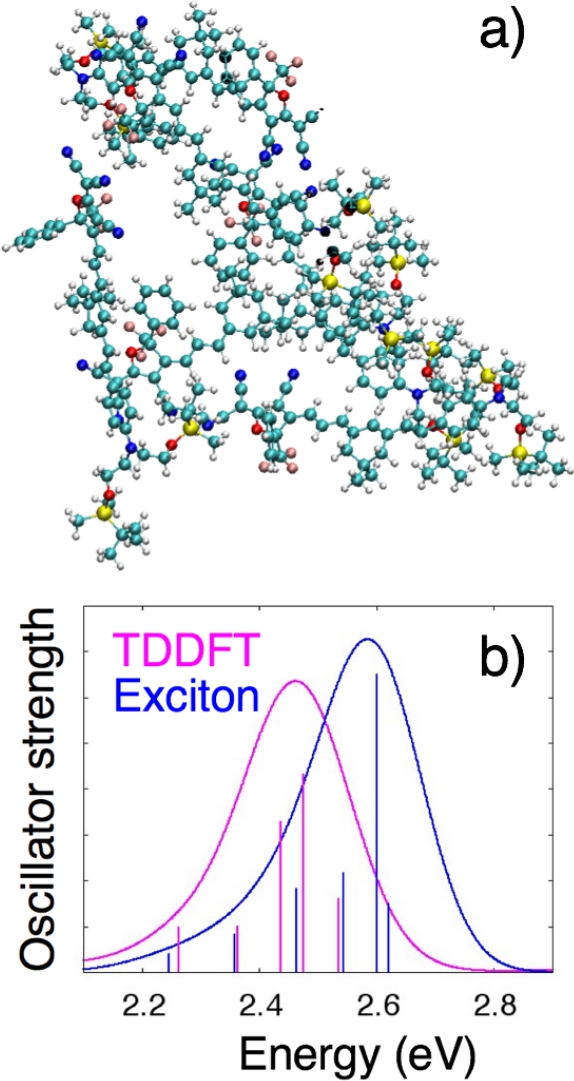
Figure 3: The structure and calculated spectrum for an aggregate of six YLD 124 molecules. (a) The aggregate structure used in the sample calculation. (b) The corresponding absorption spectra created using the exciton model Hamiltonian (blue) and an all-electron TD-DFT calculation on the entire aggregate (magenta). Please click here to view a larger version of this figure.
| 2.4458 | -0.0379 | -0.0899 | 0.0278 | -0.0251 | 0.0120 |
| -0.0379 | 2.4352 | -0.0056 | -0.1688 | -0.0070 | -0.0085 |
| -0.0899 | -0.0056 | 2.5111 | 0.0032 | 0.0239 | 0.0794 |
| 0.0278 | -0.1688 | 0.0032 | 2.3954 | 0.0057 | 0.0073 |
| -0.0251 | -0.0070 | 0.0239 | 0.0057 | 2.5171 | -0.0211 |
| 0.0120 | -0.0085 | 0.0794 | 0.0073 | -0.0211 | 2.5256 |
Table 1: The Hamiltonian for a sample calculation on the aggregate of six YLD 124 molecules shown in Figure 3a. The diagonal elements are the excitation energies of individual molecules; the off-diagonal elements are the excitonic couplings between molecules (all values are in eV).
