In den letzten zwei Jahrzehnten haben Feststoffe und Folien, die aus aggregierten organischen Molekülen hergestellt werden, mehrere Anwendungen in optoelektronischen Geräten gefunden. Geräte, die auf solchen Materialien basieren, haben viele attraktive Eigenschaften, einschließlich geringem Gewicht, Flexibilität, geringem Stromverbrauch und Potenzial für eine billige Produktion mit Inkjet-Druck. Displays, die auf organischen Leuchtdioden (OLEDs) basieren, ersetzen flüssige kristalline Displays als State-of-the-Art für Mobiltelefone, Laptops, Fernsehgeräte und andere elektronische Geräte1,2,3,4. Die Bedeutung von OLEDs für Beleuchtungsanwendungen wird in den kommenden Jahren voraussichtlich zunehmen4. Die Leistung von organischen Photovoltaik-Geräten verbessert sich stetig, mit Leistungsumwandlungseffizienzen von über 16% kürzlich für Ein-Kreuzung organische Solarzellen5gemeldet. Organische Materialien haben auch das Potenzial, andere Technologien zu stören, wie glasfaserkommunikation, wo ihre Verwendung die Entwicklung von elektro-optischen Modulatoren mit extrem hohen Bandbreiten von 15 THz und mehr6,7ermöglicht.
Eine große Herausforderung bei der Optimierung von Festkörper-Molekularmaterialien für Anwendungen in der Optoelektronik besteht darin, dass ihre Eigenschaften in der Regel stark von der nanoskaligen Struktur des Materials abhängen. Der Produktionsprozess ermöglicht die Definition der Nanostruktur eines Materials bis zu einem gewissen Grad durch den Einsatz kontrollierter Wachstumstechniken, wie chemische Dampfabscheidung,8 Verformung von optisch aktiven Molekülen auf ein anderes Material (d.h. eine Polymermatrix9,10), thermisches Glühen11,12usw. Nanoskalenstörungen sind jedoch für die meisten molekularen Materialien intrinsisch und können in der Regel nicht vollständig beseitigt werden. Daher ist es für die rationale Gestaltung organischer optoelektronischer Materialien unerlässlich, zu verstehen, wie sich Unordnung auf die Eigenschaften eines Materials auswirkt, und Wege zu finden, es für eine optimale Leistung zu entwickeln.
Der Grad der Störung in molekularen Materialien ist in der Regel zu groß, um es als Störung einer periodischen kristallinen Struktur mit einer elektronischen Struktur zu behandeln, die durch Bandtheorie beschrieben werden kann. Andererseits ist die Anzahl der Moleküle, die in eine Simulation einbezogen werden müssen, um die Eigenschaften eines Schüttguts oder eines Films zu reproduzieren, zu groß, um erste Prinzipien quantenchemischer Methoden wie Dichtefunktionstheorie (DFT)13,14 und zeitabhängige Dichtefunktionstheorie (TD-DFT)15,16zu verwenden. Organische Moleküle mit Anwendungen in der Optoelektronik haben in der Regel relativ große , konjugierte Systeme; viele haben auch Spender- und Akzeptorgruppen. Die Erfassung des korrekten Ladungsübertragungsverhaltens in solchen Molekülen ist für die Berechnung ihrer optoelektronischen Eigenschaften unerlässlich, kann aber nur mit fernkorrigierten Hybridfunktionen in TD-DFT17,18,19,20erreicht werden. Berechnungen, die solche Funktionen verwenden, skalieren super linear mit der Größe des Systems und sind derzeit nur praktisch, um die optoelektronischen Eigenschaften einzelner organischer Moleküle oder kleiner molekularer Aggregate zu modellieren, die mit nicht mehr als 104 atomaren Basisfunktionen beschrieben werden können. Eine Simulationsmethode, die ungeordnete Materialien beschreiben könnte, die aus einer großen Anzahl von Chromophoren bestehen, wäre sehr nützlich für die Modellierung dieser Systeme.
Die Größe intermolekularer Wechselwirkungen in molekularen Materialien ist oft vergleichbar oder kleiner als die Reihenfolge der Variation der energetischen Parameter (wie die Eigenstate-Energien oder Anregungsenergien) zwischen einzelnen Molekülen, aus denen das Material besteht. In solchen Fällen ist Multiskalenmodellierung der vielversprechendste Ansatz, um die optoelektronischen Eigenschaften großer ungeordneter molekularer Systeme zu verstehen und zu optimieren21,22,23. Dieser Ansatz verwendet quantenchemische Methoden erster Prinzipien (in der Regel DFT und TD-DFT), um die Eigenschaften einzelner Moleküle, aus denen das Material besteht, genau zu berechnen. Der Hamiltonian einer Materialprobe, die groß genug ist, um das Massenmolekülmaterial darzustellen (vielleicht durch die Verwendung periodischer Randbedingungen), wird dann mit den Parametern konstruiert, die für einzelne Moleküle berechnet wurden. Dieser Hamiltonian kann dann verwendet werden, um die optoelektronischen Parameter eines großen molekularen Aggregats, eines dünner Films oder eines Massenmolekülmaterials zu berechnen.
Exciton-Modelle sind eine Unterklasse von Multiskalenmodellen, in denen angeregte Zustände eines molekularen Materials auf der Basis von Exzitendargestellt werden: Elektronenlochpaare, die durch Die Coulomb-Attraktion24,25gebunden sind. Für die Modellierung vieler angeregter Zustandsprozesse reicht es aus, nur Frenkel-Exzitonen26einzubeziehen, bei denen das Elektron und das Loch auf demselben Molekül lokalisiert sind. Ladungsübertragungsexztonen, bei denen das Elektron und das Loch auf verschiedenen Molekülen lokalisiert sind, müssen in einigen Fällen (z. B. bei der Modellierung der Ladungstrennung in Spender-Akzeptor-Systemen)27,28enthalten sein. Obwohl Exciton-Modelle Multiskalenmodelle sind, die nur mit Berechnungen des ersten Prinzips an einzelnen Molekülen parametrisiert werden können, berücksichtigen sie immer noch intermolekulare Wechselwirkungen. Die beiden primären Wechselwirkungstypen, die sie berücksichtigen können, sind (a) exzitonische Kopplungen zwischen Molekülen, die die Fähigkeit von Exziten charakterisieren, sich zwischen Molekülen zu delokalisieren oder zwischen Molekülen zu übertragen, und (b) elektrostatische Polarisation der Elektronendichte auf einem Molekül durch die Ladungsverteilung auf umgebenden Molekülen. Wir haben bereits gezeigt, dass beide Faktoren wichtig für die Modellierung der optischen und elektrooptischen Eigenschaften molekularer Aggregate sind, wie z. B. die optischen Absorptionsspektren29 und die ersten hyperpolarisierbaren30.
In diesem Beitrag stellen wir ein Protokoll zur Parametrisierung von Exzitonmodellen vor, mit dem die optischen Spektren und andere optoelektronische Eigenschaften großer molekularer Aggregate und Massenmolekülmaterialien berechnet werden können. Der excitonic Hamiltonian wird angenommen, dass ein eng bindender Hamiltonian24,25,
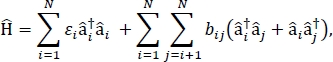
wobei i die Anregungsenergie des i.th Moleküls im Material, bij, die exzitationische Kopplung zwischen den ith- und den jj-th-Molekülen ist, sind i iund ii die Erzeugungs- bzw. Vernichtungsoperatoren für einen angeregten Zustand auf dem ith Molekül im Material.th Die exzitonic Hamiltonian Parameter werden mit TD-DFT-Berechnungen gefunden, die auf einzelnen Molekülen durchgeführt werden, aus denen das Material besteht. In diesen TD-DFT-Berechnungen wird die Ladungsverteilung auf alle anderen Moleküle im Material durch elektrostatische Einbettung von Atompunktladungen dargestellt, um die elektrostatische Polarisation der elektronischen Dichte eines Moleküls zu berücksichtigen. Die Anregungsenergien, ii, für einzelne Moleküle werden direkt aus dem TD-DFT-Berechnungsausgang entnommen. Die excitonischen Kopplungen, bij, zwischen Molekülen werden mit der Transition Density Cube Methode31berechnet, wobei die boden-zu-erregten Zustandsübergangsdichten für die interagierenden Moleküle aus der Ausgabe einer TD-DFT-Berechnung in Gauß-3232 entnommen und mit dem Multiwfn-Multifunktions-Wellenfunktionsanalysator33nachbearbeitet werden. Multiwfn Zur Simulation der Eigenschaften von Massenmolekülfeststoffen können periodische Randbedingungen auf den Hamiltonian angewendet werden.
Das aktuelle Protokoll erfordert, dass der Benutzer Zugriff auf die Programme Gaussian32 und Multiwfn33 hat. Das Protokoll wurde mit Gauß’sche 16, Revision B1 und Multiwfn Version 3.3.8 getestet, sollte aber auch für andere aktuelle Versionen dieser Programme funktionieren. Darüber hinaus verwendet das Protokoll ein benutzerdefiniertes C++-Dienstprogramm und eine Reihe von benutzerdefinierten Python 2.7- und Bash-Skripten, deren Quellcode unter der GNU General Public License (Version 3) zur https://github.com/kocherzhenko/ExcitonicHamiltonian bereitgestellt wird. Die Berechnungen sollen auf einem Computer ausgeführt werden, auf dem ein Betriebssystem aus der Unix/Linux-Familie ausgeführt wird.
