Структурные исследования атомной шкале макромолекулярных сборок, твердотельного ЯМР спектроскопии
Summary
Структуры супрамолекулярные белка сборок на атомной резолюции имеют большое значение из-за их решающую роль в различных биологических явлений. Здесь мы представляем протокол для выполнения высокого разрешения структурных исследований на нерастворимы и некристаллических высокомолекулярных белков сборок магии-угол спиннинг твердотельного ЯМР спектроскопии (MAS SSNMR).
Abstract
Супрамолекулярная белка сборки фундаментальные роли в биологических процессах, начиная от хост возбудитель взаимодействия, вирусные инфекции для распространения нейродегенеративных расстройств. Такие сборки состоят в несколько субблоков белка, организованных non ковалентные образом сформировать большой макромолекулярных объектов, которые можно выполнять различные клеточные функции или вызвать негативные последствия. Атомной понимание механизмов Ассамблеи и функционирования этих макромолекулярных сборок часто остаются скудными с их неотъемлемым нерастворимость и не кристалличности часто резко снижает качество данных, полученных из большинства методов используется в структурной биологии, например рентгеноструктурного анализа и решения ядерного магнитного резонанса (ЯМР). Здесь мы представляем магии угол спиннинг твердотельного ЯМР спектроскопии (SSNMR) как мощный метод для изучения структуры макромолекулярных сборок на атомной резолюции. SSNMR можно выявить атомной детали на собранный комплекс без ограничения размера и растворимость. Протокол, представленные здесь описываются основные шаги от производства 13C /15N меченных изотопом высокомолекулярных белков сборки на приобретение стандартных SSNMR спектры и их анализа и интерпретации. В качестве примера мы покажем трубопровода Ассамблеи нитчатые протеина структурного анализа SSNMR.
Introduction
Достижения в магии угол спиннинг твердотельного ЯМР спектроскопии (SSNMR) предлагают эффективный инструмент для структурной характеристики сборок высокомолекулярных белков в атомной резолюции. Эти сборки белков являются вездесущими систем, которые играют важную роль во многих биологических процессах. Их молекулярные структуры, взаимодействий и динамика доступны для SSNMR исследований, как уже было показано для вирусных (1capsids) и механизмы бактериальной инфекции (секреции систем2,3, пили4), мембраны белковых комплексов5,6,,78 и функциональных Амилоиды 9,10,11. Этот тип молекулярной сборки может также провоцировать патологий, таких как в нейродегенеративных заболеваний, где белки собрать в Протеолиз, амилоида государствах и вызвать поведение аномальные клетки или клетки смерти 12,13. Белка сборки часто строятся путем симметричного олигомеризации кратные копий субблоков белок в больших супрамолекулярные объектов различной формы, включая фибрилл, нитей, поры, трубы или наночастиц. Четвертичные архитектура определяется слабого взаимодействия между белковых субъединиц организовать пространственной и временной Ассамблеи и для проведения сложных биологических функций. Структурные исследования на атомной шкале на эти сборки являются вызовом для высокого разрешения методов с их внутренней нерастворимость и очень часто их не кристалличности ограничивает использование обычных рентгеноструктурного анализа или решения ЯМР подходы. Магия угол спиннинг (MAS) SSNMR является новой техникой для получения данных атомной резолюции нерастворимые высокомолекулярные сборок и доказал свою эффективность для решения 3D атомной модели для все большего числа сложных биомолекулярных систем, включая Бактериальные нитей, амилоида сборки и вирусных частиц 14,,1516,,1718,19,20, 21,22. Технические авансы на высоких магнитных полей, методологических разработок и подготовки проб создала MAS SSNMR в надежный метод расследовать нерастворимые белки в различных средах, особенно в их биологически соответствующих высокомолекулярных соединений собрал государством или в клеточных мембран, что делает технику очень дополнение к крио электронной микроскопии. Во многих случаях очень высокая степень симметрии характеризует такие сборки белков. MAS SSNMR использует эту функцию, как всех субблоков протеина в сборке homomolecular будет иметь ту же структуру местных и поэтому практически одинаковую подпись SSNMR, резко уменьшая сложность анализа.
Эффективный протокол для структурных исследований высокомолекулярных белков сборок по умеренной MAS (< 25 кГц) SSNMR представлены в этом видео и могут быть разделены на различные этапы (рис. 1). Мы продемонстрируем критических этапов рабочего SSNMR структурные исследования свидетельствуют о волокнистых белков Ассамблеи (см. выделены шаги на рис. 1), за исключением очистки субъединицы белка, различные для каждого белка Ассамблея но решающее значение для структурных исследований и не вдаваясь в технические/методологические детали SSNMR спектроскопии и структуры расчета какие специализированные учебники доступны онлайн. Хотя настоящий Протокол будет главным образом сосредоточена на твердотельного ЯМР эксперименты, проведенные в условиях MAS, использование выровнены биологических сред 23,24,25,26 , 27, таких как соответствие bicelles, позволяют для расследования конформации белка и динамических протеин взаимодействия в средствах массовой информации мембраны как без MAS технологии. Мы будем показывать выражения протеина и Ассамблее шаги, а также запись решающую спектры SSNMR и их анализа и интерпретации. Наша цель – обеспечить понимание структурного анализа трубопровода, позволяя читателю выполнить атомно резолюции структурные исследования высокомолекулярных Ассамблеи SSNMR методами.
Протокол включает в себя 3 секции:
1. твердотельного ЯМР образца производство
Предпосылкой для твердотельного ЯМР анализ, белковых компонентов макромолекулярных Ассамблеи необходимо быть выражены, меченных изотопом, очищенная и собран в пробирке в родной как комплекс состояние (для примера см. Рисунок 2) . Чтобы обеспечить высокую чувствительность ЯМР, обогащения изотопов в 13C и 15N маркировки требуется минимальный бактериальный выражение массовой информации дополнена 13C и 15N источников, таких как равномерно 13 C-меченых глюкозы/глицерина и 15NH4Cl соответственно. В поздней стадии протокола, выборочно 13C-меченых образцов производится с выборочно 13C-меченых источников, таких как (1,3 –13C)- и (2 –13C)-глицерин (или (1 –13C)- и (2 –13C)- глюкоза) используются для облегчения анализа ЯМР. Смешанные помечены образец, соответствующий эквимолярных смесь из 50% 15N – и 50% 13C-меченых или 50% (1,3 –13C)- и 50% (2 –13C)-глюкозы вводятся для описания обнаружения межмолекулярных взаимодействий. Высокая степень чистоты белка, а также жесткие условия на этапе сборки являются ключевыми факторами для обеспечения однородной структурного порядка окончательного образца.
2. Предварительный структурных характеристик на основе одномерный твердотельного ЯМР (1D)
Мы представляем основные эксперименты для структурного анализа, SSNMR. Одномерный (1D) кросс поляризация (CP) и INEPT / RINEPT28 экспериментов, обнаруженные на 13C ядер используется для обнаружения жестких и гибких белка сегментов в Ассамблее, соответственно и оценить степень структурной однородность и местные полиморфизм (для примера см. рис. 3).
Жун > 3. Конформационный анализ и 3D определения структуры
Подразделы 1 и 2 касаются конформационный анализ, который основывается на SSNMR резонанс назначение всех жестких остатки белка Ассамблеи, как химические сдвиги являются очень чувствительных зондов для местной окружающей среды и может использоваться для прогнозирования Пхи/psi двугранных углов и тем самым определить вторичную структуру. Рисунок 4 иллюстрирует пример последовательного резонанса уступки в ядре жесткой белка Ассамблеи. Определение 3D структура основано на коллекции структурных данных, такие, как ограничения расстояния кодирования закрыть proximities (< Å 7-9), содержащие внутри – и межмолекулярных информации. Подпункты 3 и 4 описывают дальнего действия далеких сдержанность сбора и интерпретации. Загрязнении контактов определяются как внутримолекулярной 13C –13C proximities вытекающих из остатков i j, с | i-j | ≥4, определяя тем самым складке третичного белка мономерных Субблок, или как межмолекулярных 13C –13C proximities, определение межмолекулярных взаимодействий между субблоков протеина в Ассамблее. Внутри – и межмолекулярных интерфейсы показано на рисунке 5. SSNMR ограничений обнаруживаются через 13C –13C и 15N –13C сцепленный эксперименты обычно кодировать для internuclear расстояния < 1 Нм. Подраздел 4 объясняет обнаружения межмолекулярных расстояние ограничений. В симметричных белка сборках, использование однородно помечены выборок (т.е. 100% равномерно или выборочно помечены) для выявления межмолекулярных Субблок Субблок взаимодействия является ограниченным, как оба интра – и Межамери – молекулярных контактов приведет к обнаружить сигналы. Недвусмысленный обнаружения межмолекулярных proximities достигается с помощью смешанных помечены образцы, содержащие эквимолярных смесь двух по-разному помечены образцов, комбинированные до агрегирования. Подраздел 5 кратко знакомит структуры моделирования.
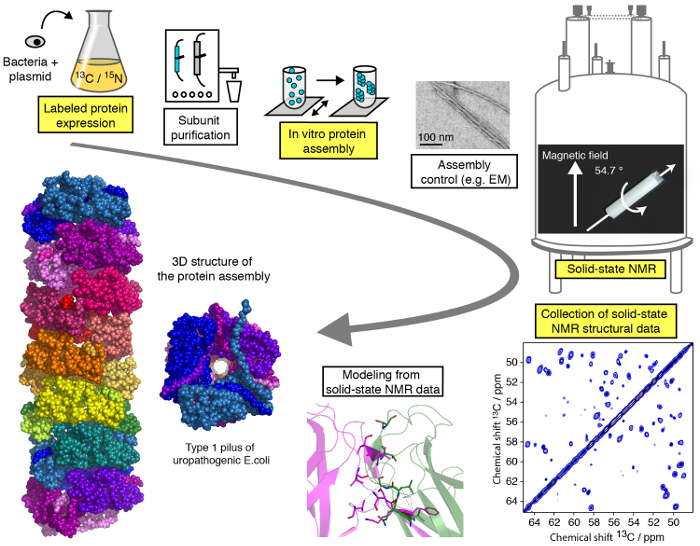
Рисунок 1 : Рабочий процесс атомно резолюции структурные исследования твердотельного ЯМР. 13 C, 15N изотоп помечены производства белка, субъединицы очистки, субъединицы Ассамблеи, контроль формирования Ассамблеи, SSNMR эксперименты, SSNMR эксперимент анализа и извлечения расстояние ограничений и показаны структура моделирования. Пожалуйста, нажмите здесь, чтобы посмотреть большую версию этой фигуры.
Protocol
Representative Results
Discussion
Твердотельного ЯМР (SSNMR) является методом выбора для характеризующие высокомолекулярных белков сборок на атомарном уровне. Одним из центральных вопросов в определении на основе SSNMR структуры является спектральной качество исследуемой системы, что позволяет установить 3D структурных моделей различных точности, обычно начиная с разрешением модели (содержащий вторичного Структура элементов и мало 3D информации) для псевдо-атомной трехмерных структур. Количество и качество структурных сведений, извлеченных из многомерного SSNMR экспериментов является ключом для вычисления структуру ЯМР высокого разрешения Ассамблеи.
Описывается протокол основывается на обнаружение 13C –13C и 15N –13C структурные ограничения требующие записи спектров несколько 2D (и иногда 3D) с высокой сигнал шум. При умеренной MAS частотах (< 25 кГц), образец вводится в роторы с размерами 3,2-4 мм диаметром, позволяя для количеств белка до ~ 50 мг, зависит от образца гидратации. Количество образцов внутри ротора прямо пропорциональна соотношение сигнал шум в SSNMR спектры, решающим фактором для обнаружения дальнего расстояния ограничений и их однозначной назначения.
Cпектральное разрешение является параметром решающее значение во время присваивания последовательный резонанс и коллекции ограничений. Для получения оптимальных результатов, параметры подготовки образца должны быть оптимизированы, особенно в очищение субъединицы и Ассамблея условий (pH, буфера, тряска, температуры и т.д.). Пример оптимизации рекомендуется подготовить немеченого образцы для нескольких отдельных условий для которого наблюдается Ассамблеи и для записи 1D 1H –13C CP спектра (описано в шаге 2.1) на каждого подготовленного образца. Спектры служат для сравнения спектральное разрешение и дисперсия между различных препаратов, на основе которой можно определить оптимальные условия.
Качество данных SSNMR сильно зависит от выбора параметров приобретение ЯМР, особенно для передачи шаги поляризации. Использование высоких магнитных прочностями поля ( 1H частота ≥600 МГц) имеет важное значение для высокой чувствительности и спектральным разрешением, требуется когда сталкиваются сложных целей таких сборок высокомолекулярных белков.
Ограничивающим фактором во многих случаях является наличие спектрометр. Поэтому разумный выбор образцов должен быть подготовлен должно предшествовать спектрометр сессии. В любом случае, равномерно 13C, 15N-меченых образца является необходимым условием для выполнения назначения последовательных и внутри остаточного резонанс. Белки, присвоенный твердотельного ЯМР методами71см. Определение структуры макромолекулярных сборок при умеренной MAS частотах требует выборочно 13C-меченых образцов; для обнаружения на большие расстояния 13C –13C и 13C –15N контакты пробы, основанные на 1,3 –13C – и 2 –13C-gylcerol или 1 –13C – и 2 –13C-глюкозы маркировки являются обычно используется, как описано выше. Выбор между двумя схемами маркировки основана на спектральные отношение сигнал шум и резолюции. Чтобы различать внутри – и межмолекулярных контактов на большие расстояния, смешанные маркировку и разбавленных образцы показали эффективным.
Короче говоря, критические шаги для атомной SSNMR структурные исследования являются: (i подготовка подразделений и Ассамблее необходимо оптимизировать получить отличный образец количество и качество, прочность и приобретения параметры (ii) спектрометр поля должны быть выбраны тщательно; (iii) селективного маркировки стратегии необходимы для определения 3D структуры и количество требуемых данных зависит от качества данных и наличия дополнительных данных.
Несмотря на его применимость к широкому спектру супрамолекулярных систем, начиная от мембранных белков homomultimeric нано объекты SSNMR часто ограничивается потребность мг количество гетерогенны помечены материала. Последние технологические разработки в ультра-быстрой SSNMR MAS (≥100 кГц) открывают проспект 1H-обнаружены ЯМР и push предел количества минимальный пример суб мг 72,,7374. Тем не менее для детального изучения структурных 13C-меченых образцы являются незаменимыми, что ограничивает применение SSNMR образцы собраны в пробирке или систем, выраженные в организмах, которые выживают на минимальный средний, где в клеток SSNMR является новым методом (для отзывов см 75,76,77,78).
Важным фактором в SSNMR приложении, чтобы получить с высоким разрешением 3D структур является спектральное разрешение: встроенные конформационные гетерогенность в сборке может ограничить спектрального анализа спектров и резолюции. Остатков конкретных 13C маркировки может в некоторых случаях обеспечивают альтернативу получить определенное расстояние информацию о стратегических остатков с целью получения структурных моделей (для последних примеров см. 79,80).
SSNMR для определения 3D структуры по-прежнему требует коллекции нескольких наборов данных с временем сбора данных часто long на сложных инструментов, в зависимости от подхода и системы нескольких дней до недель на 600-1000 МГц (частота1H) спектрометр. Таким образом доступ к спектрометра время может быть ограничивающим фактором в углубленное исследование SSNMR.
В случае homomultimeric белка сборки, приводит к SSNMR данных достаточного качества, чтобы выявить большое количество структурных ограничений, таких как в 3,,5764,70, SSNMR по-прежнему дает без доступ к микроскопические размеры. Таким образом в определении структуры SSNMR de novo homomultimeric Ассамблеи, EM или массы в длину (MPL) данных идеально дополняют SSNMR данных для получения параметров симметрии. SSNMR данных только обеспечивают атомной внутри – и межмолекулярных интерфейсов
SSNMR отлично дополняют друг друга с структурные методы, такие как EM или MPL измерения, но данные могут также прекрасно сочетается с атомных структур, полученные кристаллографии рентгеновского снимка или решения ЯМР на мутировавших или усеченных субъединиц. Все большее количество исследований можно найти в литературе, где совместно различных структурных данных позволило для определения атомного 3D моделей высокомолекулярных сборок (см. Рисунок 6 представитель примеры).
В области структурной биологии SSNMR появляется как перспективный способ изучениянерастворимый и некристаллических сборки в атомной, т.е. предоставление структурных данных уровня атомного масштаба. В этой связи, SSNMR является Кулон решение ЯМР и рентгеноструктурного анализа для молекулярной сборки, включая мембранных белков в их родной среде и белка сборок, таких как вирусный конверты, бактериальных нитей или Амилоиды, а также РНК и РНК белковых комплексов (см., например,81). Его чрезвычайно универсальным приложениям в пробирке и в контексте сотовой, например отслеживание вторичные, третичные и четвертичные структурных изменений, выявление поверхности взаимодействия с молекулами партнера на атомной шкале (например, 82) и картирование молекулярной динамики в контексте собрал комплексов, указывают важный потенциал SSNMR в будущих структурных исследований сложных биомолекулярных сборок.
| Компонент | M9 среднего |
| NaCl | 0,5 г/Л |
| KH2PO4 | 3 г/Л |
| Na2HPO4 | 6,7 г/Л |
| MgSO4 | 1 мм |
| ZnCl2 | 10 МКМ |
| FeCl3 | 1 МКМ |
| CaCl2 | 100 МКМ |
| MEM витамин смесь 100 X | 10 мл/Л |
| 13 C-глюкоза | 2 г/Л |
| 15 NH4Cl | 1 г/Л |
Таблица 1: Состав минимальной выражение среды для рекомбинантных белков производство в E. coli клетки BL21.
Disclosures
The authors have nothing to disclose.
Acknowledgements
Эта работа финансируется НРУ (13-PDOC-0017-01 B.H. и АНР-14-CE09-0020-01-а.л.), «Инвестиции в будущее» программы IdEx Бордо/CNRS (ПЗЛ 2016 в б.х.) Справочник АНР-10-IDEX-03-02 для б.х., Фонд pour la исследований сообщала ( FRM-AJE20140630090 до а.л.), FP7 программы (FP7-люди-2013-CIG до а.л.) и Совет европейских исследований (ERC) под исследовательской и инновационной программы Европейского союза по Horizon 2020 (ERC начиная Грант а.л., соглашение не 639020) и проект ” WEAKINTERACT.»
Materials
| Instruments | |||
| NMR Spectrometer (> 11.7 Tesla) | Bruker | – | |
| triple resonance MAS SSNMR probehead | Bruker | – | |
| SSNMR rotors 4mm | Bruker | K1910 | |
| Centrifuge 5804 R | Eppendorf | 5805000629 | |
| GeneQuant 1300 spectrometer | Dutscher | 28-9182-13 | |
| IGS60 INCUBATEUR HERATHERM 75 L | Dutscher | 228001 | |
| MaxQ 4450 bench top orbital shaker | Dutscher | 78376 | |
| Tube Revolver Agitator | Dutscher | 79547 | |
| sonopuls HD 3100 | Bandelin | 3680 | |
| MicroPulser electroporator | Biorad | 165-2100 | |
| mini-PROTEAN tetra cell system | Biorad | 165-8000 | |
| AKTA pure system | GE Healthcare | 29-0182-24 | |
| capillary microman M25 pipet | Gilson | F148502 | |
| Name | Company | Catalog Number | Comments |
| Materials | |||
| amiconR ultra-15 | sigma | Z740199-8EA | |
| capillaries and pistons | Gilson | F148112 | |
| spatula | Fisher | 13263799 | |
| Name | Company | Catalog Number | Comments |
| Reagents | |||
| D-glucose 13C6 | Sigma | 389374 | |
| Ammonium-15N-chloride | Sigma | 299251 | |
| 1,3 13C2 glycerol | Sigma | 492639 | |
| 2 13C glycerol | Sigma | 489484 | |
| Kanamycin | Sigma | K1876 | |
| Carbenicillin | Sigma | C3416 | |
| Sodium phosphate dibasic | Sigma | S7907 | |
| Potassium phosphate monobasic | Sigma | P5655 | |
| Sodium chloride | Sigma | 71380 | |
| calcium chloride | Sigma | C1016 | |
| Magnesium sulfate | Sigma | 208094 | |
| Iron Chloride | Sigma | 157740 | |
| Zinc chloride | Sigma | 793523 | |
| MEM Vitamin Solution (100×) | Sigma | M68954 | |
| IPTG | Fisher | BP1755 | |
| Trizma base | Sigma | T1503 | |
| Tricine | Sigma | T0377 | |
| SDS | Sigma | 436143 | |
| sodium azide | sigma | 71289 | |
| 4,4-dimethyl-4-silapentane-1-sulfonic acid | Sigma | 178837 | |
| Name | Company | Catalog Number | Comments |
| Softwares | |||
| Unicorn 6.3 | GE Healthcare | Akta systems | |
| ccpNMR | CCPN | spectrometer systems |
References
- Morag, O., Sgourakis, N. G., Baker, D., Goldbourt, A. The NMR-Rosetta capsid model of M13 bacteriophage reveals a quadrupled hydrophobic packing epitope. Proc Natl Acad Sci U S A. 112 (4), 971-976 (2015).
- Loquet, A., et al. Atomic model of the type III secretion system needle. Nature. 486 (7402), 276-279 (2012).
- Demers, J. P., et al. High-resolution structure of the Shigella type-III secretion needle by solid-state NMR and cryo-electron microscopy. Nat Commun. 5 (4976), (2014).
- Habenstein, B., et al. Hybrid Structure of the Type 1 Pilus of Uropathogenic Escherichia coli. Angew Chem Int Ed Engl. 54 (40), 11691-11695 (2015).
- Cady, S. D., et al. Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature. 463 (7281), 689-692 (2010).
- Park, S. H., et al. Structure of the chemokine receptor CXCR1 in phospholipid bilayers. Nature. 491 (7426), 779-783 (2012).
- Kaplan, M., et al. Probing a cell-embedded megadalton protein complex by DNP-supported solid-state NMR. Nat Methods. 12 (7), 649-652 (2015).
- Wang, S., et al. Solid-state NMR spectroscopy structure determination of a lipid-embedded heptahelical membrane protein. Nat Methods. 10 (10), 1007-1012 (2013).
- Daskalov, A., et al. Signal transduction by a fungal NOD-like receptor based on propagation of a prion amyloid fold. PLoS Biol. 13 (2), e1002059 (2015).
- Daskalov, A., et al. Identification of a novel cell death-inducing domain reveals that fungal amyloid-controlled cell death is related to necroptosis. Proc Natl Acad Sci U S A. , (2016).
- Li, J., et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell. 150 (2), 339-350 (2012).
- Knowles, T. P., Vendruscolo, M., Dobson, C. M. The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol. 15 (6), 384-396 (2014).
- Aguzzi, A., Lakkaraju, A. K. Cell Biology of Prions and Prionoids: A Status Report. Trends Cell Biol. 26 (1), 40-51 (2016).
- Habenstein, B., Loquet, A. Solid-state NMR: An emerging technique in structural biology of self-assemblies. Biophys Chem. , (2015).
- Meier, B. H., Bockmann, A. The structure of fibrils from ‘misfolded’ proteins. Curr Opin Struct Biol. 30, 43-49 (2015).
- Miao, Y., Cross, T. A. Solid state NMR and protein-protein interactions in membranes. Curr Opin Struct Biol. 23 (6), 919-928 (2013).
- Tang, M., Comellas, G., Rienstra, C. M. Advanced solid-state NMR approaches for structure determination of membrane proteins and amyloid fibrils. Acc Chem Res. 46 (9), 2080-2088 (2013).
- Weingarth, M., Baldus, M. Solid-state NMR-based approaches for supramolecular structure elucidation. Acc Chem Res. 46 (9), 2037-2046 (2013).
- Loquet, A., Habenstein, B., Lange, A. Structural investigations of molecular machines by solid-state NMR. Acc Chem Res. 46 (9), 2070-2079 (2013).
- Yan, S., Suiter, C. L., Hou, G., Zhang, H., Polenova, T. Probing structure and dynamics of protein assemblies by magic angle spinning NMR spectroscopy. Acc Chem Res. 46 (9), 2047-2058 (2013).
- Tycko, R., Wickner, R. B. Molecular structures of amyloid and prion fibrils: consensus versus controversy. Acc Chem Res. 46 (7), 1487-1496 (2013).
- Hong, M., Zhang, Y., Hu, F. Membrane protein structure and dynamics from NMR spectroscopy. Annu Rev Phys Chem. 63, 1-24 (2012).
- Jelinek, R., Ramamoorthy, A., Opella, S. J. High-Resolution Three-Dimensional Solid-state NMR Spectroscopy of a Uniformly 15N-Labeled Protein. J Am Chem Soc. 117, 12348-12349 (1995).
- Xu, J., et al. Bicelle-enabled structural studies on a membrane-associated cytochrome B5 by solid-state MAS NMR spectroscopy. Angew Chem Int Ed Engl. 47 (41), 7864-7867 (2008).
- Durr, U. H., Gildenberg, M., Ramamoorthy, A. The magic of bicelles lights up membrane protein structure. Chem Rev. 112 (11), 6054-6074 (2012).
- Yamamoto, K., et al. Probing the transmembrane structure and topology of microsomal cytochrome-p450 by solid-state NMR on temperature-resistant bicelles. Sci Rep. 3, 2556 (2013).
- Huang, R., et al. Probing the transmembrane structure and dynamics of microsomal NADPH-cytochrome P450 oxidoreductase by solid-state NMR. Biophys J. 106 (10), 2126-2133 (2014).
- Durr, U. H., Yamamoto, K., Im, S. C., Waskell, L., Ramamoorthy, A. Solid-state NMR reveals structural and dynamical properties of a membrane-anchored electron-carrier protein, cytochrome b5. J Am Chem Soc. 129 (21), 6670-6671 (2007).
- Hong, M. Determination of multiple phi-torsion angles in proteins by selective and extensive (13)C labeling and two-dimensional solid-state NMR. J Magn Reson. 139 (2), 389-401 (1999).
- Lundstrom, P., et al. Fractional 13C enrichment of isolated carbons using [1-13C]- or [2- 13C]-glucose facilitates the accurate measurement of dynamics at backbone Calpha and side-chain methyl positions in proteins. J Biomol NMR. 38 (3), 199-212 (2007).
- Loquet, A., Lv, G., Giller, K., Becker, S., Lange, A. 13C spin dilution for simplified and complete solid-state NMR resonance assignment of insoluble biological assemblies. J Am Chem Soc. 133 (13), 4722-4725 (2011).
- Castellani, F., et al. Structure of a protein determined by solid-state magic-angle-spinning NMR spectroscopy. Nature. 420 (6911), 98-102 (2002).
- Higman, V. A., et al. Assigning large proteins in the solid state: a MAS NMR resonance assignment strategy using selectively and extensively 13C-labelled proteins. J Biomol NMR. 44 (4), 245-260 (2009).
- Bockmann, A., et al. Characterization of different water pools in solid-state NMR protein samples. J Biomol NMR. 45 (3), 319-327 (2009).
- Cavanagh, J., Fairbrother, W. J., Palmer, A. G., Skelton, N. J. . Protein NMR spectroscopy, principles and practice. , (1996).
- Hartman, S. R., Hahn, E. L. Nuclear Double Resonance in the Rotating Frame. Phys Rev. 128 (5), 2042-2053 (1962).
- Harris, R. K., et al. Further conventions for NMR shielding and chemical shifts IUPAC recommendations 2008. Solid State Nucl Magn Reson. 33 (3), 41-56 (2008).
- Wang, Y., Jardetzky, O. Probability-based protein secondary structure identification using combined NMR chemical-shift data. Protein Sci. 11 (4), 852-861 (2002).
- Shaka, A. J., Baker, P. B., Freeman, R. Computer-Optimized Scheme for Wideband Applications and Low-Level Operation. J Magn Reson. 64, 547-552 (1985).
- Szeverenyi, N. M., Sullivan, M. J., Maciel, G. E. Observation of Spin Exchange by Two-Dimensional Fourier-Transform C-13 Cross Polarization-Magic-Angle Spinning. J Magn Reson. 47, 462-475 (1982).
- Baldus, M., Petkova, A. T., Herzfeld, J., Griffin, R. G. Cross polarization in the tilted frame: assignment and spectral simplification in heteronuclear spin systems. Mol Phys. 95 (5), 1197-1207 (1998).
- Verel, R., Ernst, M., Meier, B. H. Adiabatic dipolar recoupling in solid-state NMR: the DREAM scheme. J Magn Reson. 150 (1), 81-99 (2001).
- . Sparky – NMR Assignment and Integration Software Available from: https://www.cgl.ucsf.edu/home/sparky/ (2017)
- Luca, S., et al. Secondary chemical shifts in immobilized peptides and proteins: a qualitative basis for structure refinement under magic angle spinning. J Biomol NMR. 20 (4), 325-331 (2001).
- Shen, Y., Bax, A. SPARTA+: a modest improvement in empirical NMR chemical shift prediction by means of an artificial neural network. J Biomol NMR. 48 (1), 13-22 (2010).
- Berjanskii, M. V., Neal, S., Wishart, D. S. PREDITOR: a web server for predicting protein torsion angle restraints. Nucleic Acids Res. 34 (Web Server issue), W63-W69 (2006).
- Bardiaux, B., Malliavin, T., Nilges, M. ARIA for solution and solid-state NMR. Methods Mol Biol. 831, 453-483 (2012).
- Guerry, P., Herrmann, T. Comprehensive automation for NMR structure determination of proteins. Methods Mol Biol. 831, 429-451 (2012).
- Vasa, S., et al. beta-Helical architecture of cytoskeletal bactofilin filaments revealed by solid-state NMR. Proc Natl Acad Sci U S A. 112 (2), E127-E136 (2015).
- He, L., et al. Structure determination of helical filaments by solid-state NMR spectroscopy. Proc Natl Acad Sci U S A. 113 (3), E272-E281 (2016).
- Tang, M., et al. High-resolution membrane protein structure by joint calculations with solid-state NMR and X-ray experimental data. J Biomol NMR. 51 (3), 227-233 (2011).
- Paravastu, A. K., Leapman, R. D., Yau, W. M., Tycko, R. Molecular structural basis for polymorphism in Alzheimer’s beta-amyloid fibrils. Proc Natl Acad Sci U S A. 105 (47), 18349-18354 (2008).
- Schutz, A. K., et al. Atomic-resolution three-dimensional structure of amyloid beta fibrils bearing the Osaka mutation. Angew Chem Int Ed Engl. 54 (1), 331-335 (2015).
- Sgourakis, N. G., Yau, W. M., Qiang, W. Modeling an in-register, parallel "iowa" abeta fibril structure using solid-state NMR data from labeled samples with rosetta. Structure. 23 (1), 216-227 (2015).
- Lewandowski, J. R., De Paepe, G., Griffin, R. G. Proton assisted insensitive nuclei cross polarization. J Am Chem Soc. 129 (4), 728-729 (2007).
- Carlon, A., et al. How to tackle protein structural data from solution and solid state: An integrated approach. Prog Nucl Magn Reson Spectrosc. 92-93, 54-70 (2016).
- Judge, P. J., Taylor, G. F., Dannatt, H. R., Watts, A. Solid-state nuclear magnetic resonance spectroscopy for membrane protein structure determination. Methods Mol Biol. 1261, 331-347 (2015).
- Wang, S., Ladizhansky, V. Recent advances in magic angle spinning solid state NMR of membrane proteins. Prog Nucl Magn Reson Spectrosc. 82, 1-26 (2014).
- Sborgi, L., et al. Structure and assembly of the mouse ASC inflammasome by combined NMR spectroscopy and cryo-electron microscopy. Proc Natl Acad Sci U S A. 112 (43), 13237-13242 (2015).
- Loquet, A., et al. Atomic structure and handedness of the building block of a biological assembly. J Am Chem Soc. 135 (51), 19135-19138 (2013).
- Walti, M. A., et al. Atomic-resolution structure of a disease-relevant Abeta(1-42) amyloid fibril. Proc Natl Acad Sci U S A. 113 (34), E4976-E4984 (2016).
- Colvin, M. T., et al. Atomic Resolution Structure of Monomorphic Abeta42 Amyloid Fibrils. J Am Chem Soc. 138 (30), 9663-9674 (2016).
- Xiao, Y., et al. Abeta(1-42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer’s disease. Nat Struct Mol Biol. 22 (6), 499-505 (2015).
- Tuttle, M. D., et al. Solid-state NMR structure of a pathogenic fibril of full-length human alpha-synuclein. Nat Struct Mol Biol. 23 (5), 409-415 (2016).
- Wasmer, C., et al. Amyloid fibrils of the HET-s(218-289) prion form a beta solenoid with a triangular hydrophobic core. Science. 319 (5869), 1523-1526 (2008).
- Van Melckebeke, H., et al. Atomic-resolution three-dimensional structure of HET-s(218-289) amyloid fibrils by solid-state NMR spectroscopy. J Am Chem Soc. 132 (39), 13765-13775 (2010).
- Lamley, J. M., et al. Solid-state NMR of a protein in a precipitated complex with a full-length antibody. J Am Chem Soc. 136 (48), 16800-16806 (2014).
- Agarwal, V., et al. De novo 3D structure determination from sub-milligram protein samples by solid-state 100 kHz MAS NMR spectroscopy. Angew Chem Int Ed Engl. 53 (45), 12253-12256 (2014).
- Stanek, J., et al. NMR Spectroscopic Assignment of Backbone and Side-Chain Protons in Fully Protonated Proteins: Microcrystals, Sedimented Assemblies, and Amyloid Fibrils. Angew Chem Int Ed Engl. 55 (50), 15504-15509 (2016).
- Baker, L. A., Baldus, M. Characterization of membrane protein function by solid-state NMR spectroscopy. Curr Opin Struct Biol. 27, 48-55 (2014).
- Luchinat, E., Banci, L. In-cell NMR: a topical review. IUCrJ. 4 (Pt 2), 108-118 (2017).
- Freedberg, D. I., Selenko, P. Live cell NMR. Annu Rev Biophys. 43, 171-192 (2014).
- Selenko, P., Wagner, G. Looking into live cells with in-cell NMR spectroscopy. J Struct Biol. 158 (2), 244-253 (2007).
- Qiang, W., Yau, W. M., Luo, Y., Mattson, M. P., Tycko, R. Antiparallel beta-sheet architecture in Iowa-mutant beta-amyloid fibrils. Proc Natl Acad Sci U S A. 109 (12), 4443-4448 (2012).
- Bateman, D. A., Tycko, R., Wickner, R. B. Experimentally derived structural constraints for amyloid fibrils of wild-type transthyretin. Biophys J. 101 (10), 2485-2492 (2011).
- Marchanka, A., Simon, B., Althoff-Ospelt, G., Carlomagno, T. RNA structure determination by solid-state NMR spectroscopy. Nat Commun. 6, 7024 (2015).
- Schutz, A. K., et al. The amyloid-Congo red interface at atomic resolution. Angew Chem Int Ed Engl. 50 (26), 5956-5960 (2011).
