Studi strutturali su scala atomica degli assembly macromolecolari di spettroscopia a risonanza magnetica di nucleare allo stato solido
Summary
Strutture di assiemi di proteina supramolecolari a risoluzione atomica sono di grande rilevanza a causa della loro un ruolo cruciale in una varietà di fenomeni biologici. Qui, presentiamo un protocollo per eseguire studi strutturali ad alta risoluzione su assiemi di proteina insolubile e non cristallini macromolecolari di magia-angolo filatura spettroscopia a risonanza magnetica di nucleare allo stato solido (SSNMR MAS).
Abstract
Gli assembly supramolecolari proteina giocano un ruolo fondamentale nei processi biologici che vanno dall’interazione ospite-patogeno, infezione virale alla propagazione di malattie neurodegenerative. Tali assemblee consistono in più subunità proteiche organizzata in un modo non-covalenti per formare grandi oggetti macromolecolari che possono eseguire una varietà di funzioni cellulari o causare conseguenze dannose. Intuizioni atomici dei meccanismi di assemblaggio e il funzionamento di tali assembly macromolecolari spesso rimangono scarse dal loro insolubilità intrinseca e non-cristallinità spesso drasticamente riduce la qualità dei dati ottenuti dalla maggior parte delle tecniche utilizzato in biologia strutturale, come la cristallografia a raggi x e risonanza magnetica nucleare (NMR) soluzione. Presentiamo qui la spettroscopia NMR allo stato solido di angolo di magia filatura (SSNMR) come un potente metodo per indagare strutture macromolecolari assembly a risoluzione atomica. SSNMR può rivelare dettagli atomici del complesso assemblato senza limitazioni di dimensioni e solubilità. Il protocollo presentato qui descrive i passi essenziali dalla produzione di 13C / assembly proteina macromolecolari isotopo-labeled15N per l’acquisizione di spettri SSNMR standard e loro analisi e interpretazione. Ad esempio, mostriamo la pipeline di un’analisi strutturale di SSNMR di un assemblaggio di proteine filamentose.
Introduction
I progressi nella magia-angolo filatura spettroscopia a risonanza magnetica di nucleare allo stato solido (SSNMR) offrono un efficace strumento per la caratterizzazione strutturale delle assemblee di proteina macromolecolari a una risoluzione atomica. Questi assembly di proteina sono onnipresenti sistemi che svolgono un ruolo essenziale in molti processi biologici. Le strutture molecolari, interazioni e dinamiche sono accessibili dagli studi SSNMR, come è stato dimostrato per virale (capsidi1) e meccanismi di infezione batterica (secrezione sistemi2,3, pili4), membrana proteina complessi5,6,7,8 e funzionale amiloidi 9,10,11. Questo tipo di assemblaggio molecolare può anche provocare patologie come ad esempio nelle malattie neurodegenerative dove proteine assemblare negli stati misfolded, amiloide e causano un comportamento aberrante delle cellule o delle cellule morte 12,13. Assembly di proteine sono spesso costruiti dall’oligomerizzazione simmetrica di copie multiple di subunità proteiche in oggetti grandi supramolecolari di varie forme tra cui fibrille, filamenti, pori, tubi o nanoparticelle. L’architettura quaternario è definita da interazioni deboli tra subunità proteiche per organizzare l’Assemblea spazio e temporale e per consentire sofisticate funzioni biologiche. Indagini strutturali scala atomica su questi assembly sono una sfida per le tecniche ad alta risoluzione dal loro insolubilità intrinseca e molto spesso loro cristallinità-non limita l’uso di convenzionale cristallografia a raggi x o soluzione NMR si avvicina. Angolo di magia filatura SSNMR (MAS) è una tecnica emergente per ottenere dati di risoluzione atomica su assembly macromolecolari insolubile e ha dimostrato la sua efficacia per risolvere modelli 3D atomiche per un numero crescente di sistemi biomolecolari complessi tra cui filamenti batterici, assemblee dell’amiloide e particelle virali 14,15,16,17,18,19,20, 21,22. Progressi tecnici su alti campi magnetici, sviluppi metodologici e preparazione del campione ha stabilito SSNMR MAS in un robusto metodo per studiare le proteine insolubili in vari ambienti, in particolare nel loro biologicamente rilevanti macromolecolari assemblati statali o nelle membrane cellulari, rendendo la tecnica altamente complementari per cryo-microscopia elettronica. In molti casi, un altissimo grado di simmetria caratterizza tali assemblee di proteina. MAS SSNMR sfrutta questa caratteristica, come tutte le unità secondarie della proteina in un assembly di inerti avrebbe la stessa struttura locale e quindi praticamente la stessa firma SSNMR, riducendo drasticamente la complessità dell’analisi.
Un efficiente protocollo per studi strutturali delle assemblee di proteina macromolecolari di MAS moderata (< 25 kHz) SSNMR è presentato in questo video e può essere suddivisa in diverse fasi (Figura 1). Mostreremo le fasi critiche del flusso di lavoro di uno studio strutturale SSNMR esemplificato su un assembly di proteine filamentose (Vedi evidenziato i passaggi nella Figura 1), con l’eccezione di purificazione della proteina di subunità, differenti per ogni proteina Assemblea ma di fondamentale importanza per gli studi strutturali e senza entrare nei dettagli tecnici/metodologico di calcolo di spettroscopia e struttura SSNMR per quale tutorial specializzati sono disponibili online. Mentre il presente protocollo si concentrerà principalmente su esperimenti NMR allo stato solido eseguiti in condizioni di MAS, l’uso di allineati ambienti biologici 23,24,25,26 , 27, ad esempio bicelles allineato, consentire per l’indagine di conformazione della proteina e interazione dinamica proteina-proteina membrana-come media senza tecnologia MAS. Vi mostreremo l’espressione della proteina e fasi di montaggio, nonché la registrazione di spettri SSNMR cruciali e loro analisi e interpretazione. Il nostro obiettivo è quello di fornire le comprensioni nella pipeline di analisi strutturale che permette al lettore di eseguire uno studio strutturale di risoluzione atomica di un assembly macromolecolare mediante tecniche di SSNMR.
Il protocollo comprende 3 sezioni:
1. la produzione del campione NMR a stato solido
Come un prerequisito per un’analisi NMR allo stato solido, le componenti proteiche della necessità di essere espressa assemblaggio macromolecolare, isotopo-etichettati, purificata e assemblati in vitro nello stato nativo-come complesso (per vedere un esempio nella figura 2) . Per garantire alta sensibilità di NMR, arricchimento dell’isotopo 13C e 15N etichettatura è necessario attraverso l’uso di media minima espressione batterica completati con 13C e 15N fonti, come uniformemente 13 C-etichetta glucosio/glicerolo e 15NH4Cl rispettivamente. Nella fase successiva del protocollo, selettivamente 13C-etichetta campioni prodotto con selettivamente 13fonti C-etichettato come (1,3 –13C)- e (2 –13C)-glicerolo (o (1 –13C)- e (2 –13C)- glucosio) vengono utilizzati per facilitare l’analisi NMR. Misto con etichettato campione corrispondente una miscela equimolare di 50% 13C-etichetta o 50% e 50% 15N – (1,3 –13C)- e il 50% (2 –13C)-glucosio sono stati introdotti per descrivere l’individuazione di intermolecolari interazioni farmacologiche. Un elevato grado di purezza della proteina, nonché a rigorose condizioni durante la fase di assemblaggio sono fattori chiave per assicurare un ordine strutturale omogeneo del campione finale.
2. preliminare caratterizzazione strutturale basata su NMR allo stato solido di unidimensionale (1D)
Vi presentiamo gli esperimenti essenziali per un’analisi strutturale di SSNMR. Unidimensionale (1D) cross-polarizzazione (CP) e inetto / RINEPT28 gli esperimenti, rilevati su nuclei di 13C sono utilizzati per rilevare segmenti rigidi e flessibili della proteina nell’Assemblea, rispettivamente e di stimare il grado di strutturale omogeneità e polimorfismo locale (per vedere un esempio nella figura 3).
Rong > 3. Determinazione della struttura analisi conformazionale e 3D
Paragrafi 1 e 2 riguardano l’analisi conformazionale, che si basa sull’assegnazione di risonanza del SSNMR rigida tutti i residui dell’Assemblea della proteina, come gli spostamenti chimici sono sonde molto sensibile all’ambiente locale e possono essere usati per predire il phi/psi diedro angoli e quindi determinare la struttura secondaria. La figura 4 illustra un esempio di un’assegnazione di risonanza sequenziale nell’anima rigida di un assemblaggio di proteine. La determinazione della struttura 3D è basata sulla raccolta di dati strutturali, ad esempio vincoli di distanza codifica chiudere vicinanze (< Å 7-9), che contiene sia intra – e intermolecolari informazioni. Interpretazione e a lungo raggio ritenuta distante collezione descrivono sottosezioni 3 e 4. Contatti a lungo raggio sono definiti come intramolecolare 13C –13C vicinanze derivanti da residui da i a j, con | i-j | ≥ 4, definendo quindi la piega terziaria della proteina dell’unità secondaria monomerica, o come intermolecolari 13C –13C vicinanze, definendo le interfacce intermolecolari tra subunità proteiche nell’assembly. Intra – e intermolecolari interfacce sono illustrate nella Figura 5. Vincoli SSNMR rilevato attraverso 13C –13C e 15N –13C accoppiamento esperimenti solitamente codificano per distanze internuclear < 1 nm. Sottosezione 4 spiega l’individuazione dei vincoli di distanza intermolecolare. Negli assembly di proteina simmetrico, l’uso di campioni etichettati in modo omogeneo (cioè 100% uniformemente o selettivamente con l’etichetta) per identificare interazioni intermolecolari subunità-unità secondaria è limitato, come entrambi intra – e inter – molecolari contatti portano a segnali rilevabili. L’individuazione univoca di vicinanze intermolecolari è ottenuta utilizzando campioni etichettati misti, contenenti una miscela equimolare di due campioni etichettati in modo diverso, combinato prima dell’aggregazione. Sottosezione 5 introduce brevemente la modellazione della struttura.
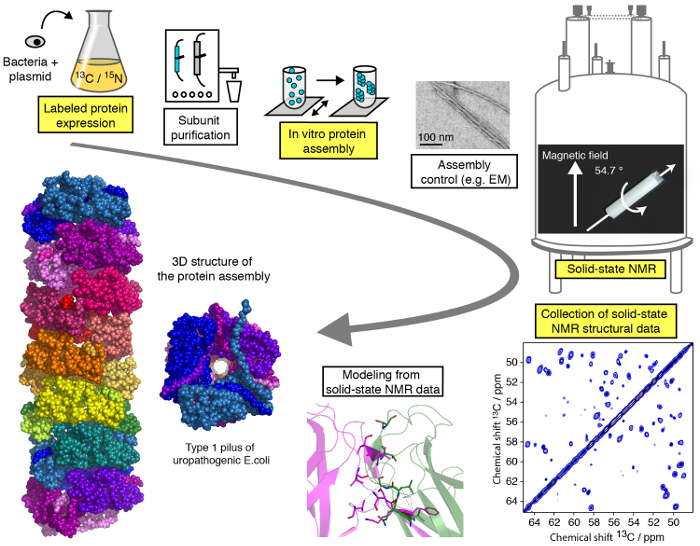
Figura 1 : Flusso di lavoro di uno studio strutturale di risoluzione atomica di NMR allo stato solido. 13 C, 15N isotopo etichettato produzione di proteine, purificazione di subunità, assemblaggio di subunità, controllo della formazione di assemblaggio, esperimenti SSNMR, SSNMR esperimento analisi e l’estrazione dei vincoli di distanza e modellazione della struttura sono indicati. Clicca qui per visualizzare una versione più grande di questa figura.
Protocol
Representative Results
Discussion
NMR allo stato solido (SSNMR) è un metodo di scelta per caratterizzare gli assembly macromolecolare della proteina a livello atomico. Uno dei temi centrali nella determinazione della struttura di base di SSNMR è la qualità spettrale del sistema oggetto dell’inchiesta, che permette di stabilire modelli strutturali 3D di precisione vari, in genere che vanno dai modelli a bassa risoluzione (contenente il secondario struttura elementi e poche informazioni 3D) per strutture 3D pseudo-atomiche. La quantità e la qualità di informazioni strutturali estratte dal multi-dimensionale SSNMR esperimenti è la chiave per calcolare una struttura NMR ad alta risoluzione dell’Assemblea.
Il protocollo descritto si basa sul rilevamento di 13C –13C e 15N –13C strutturali vincoli che richiedono la registrazione di diversi spettri 2D (e a volte 3D) con alto segnale-rumore. Alle frequenze di MAS moderate (< 25 kHz), il campione è stato introdotto in rotori con dimensioni di 3.2-4 mm di diametro permettendo per le quantità di proteine fino a ~ 50 mg, dipende l’idratazione del campione. La quantità di campione all’interno del rotore è direttamente proporzionale il rapporto segnale-rumore negli spettri di SSNMR, un fattore decisivo per l’individuazione dei vincoli di distanza a lungo raggio e loro assegnazione univoca.
La risoluzione spettrale è un parametro cruciale durante l’assegnazione di risonanza sequenziale e l’insieme di vincoli. Per ottenere risultati ottimali, i parametri di preparazione del campione devono essere ottimizzate, in particolare nella purificazione della subunità e le condizioni di montaggio (pH, buffer, agitazione, temperatura, ecc.). Per l’ottimizzazione del campione, si consiglia di preparare campioni senza etichetta per diverse circostanze distinte per i quali è stato osservato Assemblea e di registrare un 1D 1H –13C CP spettro (descritto al punto 2.1) su ogni campione preparato. Gli spettri servono per confrontare la risoluzione spettrale e dispersione tra le diverse preparazioni, basata che possono essere determinate le condizioni ottimali.
La qualità dei dati SSNMR dipende fortemente la scelta dei parametri di acquisizione NMR, soprattutto per le operazioni di trasferimento di polarizzazione. L’uso di alta intensità di campo magnetico (≥ 600 megahertz di frequenza di 1H) è essenziale per alta sensibilità e risoluzione spettrale, obbligatorio quando affrontare obiettivi complessi come gli assembly macromolecolare della proteina.
Un fattore limitante in molti casi è la disponibilità di spettrometro. Di conseguenza, una scelta giudiziosa dei campioni per essere preparati deve precedere la sessione di spettrometro. In ogni caso, un uniformemente 13C, 15N-etichettato campione è un prerequisito per eseguire l’assegnazione di risonanza sequenziale e intra-residuo. Per proteine assegnate mediante tecniche NMR allo stato solido vedere71. Determinazione della struttura delle assemblee macromolecolari a frequenze MAS moderate in modo selettivo richiede 13marcato C campioni; per la rilevazione di lungo raggio 13C –13C e 13C –15N contatti campioni basati su 1,3 –13C – e 2 –13C-gylcerol e/o 1 –13C – e 2 –13C-glucosio etichettatura sono comunemente utilizzati, come descritto in precedenza. La scelta tra i due schemi di etichettatura è basata sul rapporto segnale-rumore spettrale e risoluzione. Per distinguere tra intra – e intermolecolari contatti a lungo raggio, i campioni etichettati e diluiti misti hanno rivelato efficiente.
In breve, i passaggi critici per uno studio strutturale di SSNMR atomico sono: (i) la preparazione di subunità e la necessità di montaggio di essere ottimizzato per ottenere quantità di campione eccellente e qualità, (ii) spettrometro campo forza e acquisizione parametri devono essere scelti con cura; (iii) selettive strategie d’etichettatura sono necessari per una determinazione della struttura 3D e la quantità di dati richiesti dipende dalla qualità dei dati e la disponibilità di dati complementari.
Nonostante la sua applicabilità ad una vasta gamma di sistemi supramolecolari da proteine di membrana a homomultimeric nano-oggetti, SSNMR è spesso limitata dalla necessità per mg-quantità di materiale isotopicamente etichettato. I recenti sviluppi tecnologici nel ultra-veloce MAS (≥ 100 kHz) SSNMR aperta fino all’avenue a 1H-rilevato NMR e spingere il limite della quantità minima di campione a sub-mg 72,73,74. Tuttavia, per gli studi strutturali dettagliati 13C-etichetta campioni sono indispensabili, che limita l’applicazione di SSNMR a campioni assemblati in vitro o sistemi espressi negli organismi che sopravvivono in terreno minimo, dove nella cella SSNMR è un metodo di emergente (per recensioni Vedi 75,76,77,78).
Un fattore importante nell’applicazione di SSNMR per ottenere strutture 3D ad alta risoluzione è la risoluzione spettrale: intrinseca eterogeneità conformazionale in un assembly può limitare l’analisi spettrale spettri e risoluzione. Residuo specifico 13C etichettatura può in alcuni casi fornire un’alternativa per ottenere informazioni di distanza specifica sui residui strategici al fine di ottenere modelli strutturali (per una recente esempi Vedi 79,80).
SSNMR per determinazione della struttura 3D richiede ancora la raccolta di diversi set di dati con tempi di raccolta di dati spesso lunghi su strumenti sofisticati, a seconda l’approccio e il sistema parecchi giorni alle settimane su un 600-1000 MHz (frequenza1H) spettrometro. Di conseguenza, l’accesso al tempo spettrometro può essere un fattore limitante in un approfondito studio SSNMR.
Nel caso di assemblee di proteina di homomultimeric, che conduce ai dati SSNMR di qualità sufficiente per identificare un elevato numero di vincoli strutturali come in 3,57,64,70, SSNMR non dà ancora nessun accesso alle dimensioni microscopiche. Pertanto, in una determinazione della struttura SSNMR de novo di un assembly homomultimeric, EM o dati (MPL) massa-a-lunghezza ideale completano dati SSNMR per derivare i parametri di simmetria. SSNMR dati da soli forniscono l’atomica intra – e intermolecolari interfacce
SSNMR è altamente complementare con tecniche strutturali quali misure EM o MPL ma i dati possono essere combinati anche perfettamente con strutture atomiche ottenute mediante cristallografia a raggi x o soluzione NMR su subunità mutata o troncata. Un numero crescente di studi può essere trovato in letteratura dove la congiunzione di diversi dati strutturali ha permesso per la determinazione di modelli atomici 3D di macromolecolare assembly (vedere la Figura 6 per esempi rappresentativi).
Nel campo della biologia strutturale, SSNMR emerge come una tecnica promettente per lo studioassembly insolubili e non cristallina presso l’atomico livello, cioè fornendo dati strutturali su scala atomica. A questo proposito, SSNMR è la sospensione per soluzione NMR e cristallografia a raggi x per gli assembly molecolari, tra cui proteine di membrana nelle assemblee ambiente e proteina native come buste virale, batteriche filamenti o amiloidi, nonché di RNA e Complessi RNA-proteina (Vedi ad esempio81). Sue applicazioni altamente versatile in vitro e nel contesto cellulare, come il rilevamento delle modifiche strutturali secondarie, terziarie e quaternarie, identificando le superfici di interazione con molecole di partner su scala atomica (ad esempio 82) e mappatura dinamica molecolare nell’ambito di complessi montati, indicano il potenziale importante di SSNMR in futuri studi strutturali sugli assiemi complessi biomolecolari.
| Componente | M9 medio |
| NaCl | 0,5 g/L |
| KH2PO4 | 3 g/L |
| Na2HPO4 | 6,7 g/L |
| MgSO4 | 1 mM |
| ZnCl2 | 10 ΜM |
| FeCl3 | 1 ΜM |
| CaCl2 | 100 ΜM |
| MEM vitamina mix 100 X | 10 mL/L |
| 13 C-glucosio | 2 g/L |
| 15 NH4Cl | 1 g/L |
Tabella 1: Composizione del mezzo di espressione minima per proteina ricombinante produzione in Escherichia coli cellule BL21.
Disclosures
The authors have nothing to disclose.
Acknowledgements
Questo lavoro è finanziato dalla ANR (13-PDOC-0017-01 B.H. e ANR-14-CE09-0020-01 a A.L.), “Gli investimenti per il futuro” programma IdEx Bordeaux/CNRS (PEPS 2016 al B.H.) fare riferimento ANR-10-IDEX-03-02 al B.H., la Fondation pour la Recherche Médicale ( FRM-AJE20140630090 a A.L.), il programma del 7 ° PQ (7 ° PQ-persone-2013-CIG a A.L.) e del Consiglio europeo della ricerca (CER) nell’ambito del programma dell’Unione europea Horizon 2020, ricerca e innovazione (ERC Starting Grant di A.L., accordo No 639020) e progetto ” WEAKINTERACT.”
Materials
| Instruments | |||
| NMR Spectrometer (> 11.7 Tesla) | Bruker | – | |
| triple resonance MAS SSNMR probehead | Bruker | – | |
| SSNMR rotors 4mm | Bruker | K1910 | |
| Centrifuge 5804 R | Eppendorf | 5805000629 | |
| GeneQuant 1300 spectrometer | Dutscher | 28-9182-13 | |
| IGS60 INCUBATEUR HERATHERM 75 L | Dutscher | 228001 | |
| MaxQ 4450 bench top orbital shaker | Dutscher | 78376 | |
| Tube Revolver Agitator | Dutscher | 79547 | |
| sonopuls HD 3100 | Bandelin | 3680 | |
| MicroPulser electroporator | Biorad | 165-2100 | |
| mini-PROTEAN tetra cell system | Biorad | 165-8000 | |
| AKTA pure system | GE Healthcare | 29-0182-24 | |
| capillary microman M25 pipet | Gilson | F148502 | |
| Name | Company | Catalog Number | Comments |
| Materials | |||
| amiconR ultra-15 | sigma | Z740199-8EA | |
| capillaries and pistons | Gilson | F148112 | |
| spatula | Fisher | 13263799 | |
| Name | Company | Catalog Number | Comments |
| Reagents | |||
| D-glucose 13C6 | Sigma | 389374 | |
| Ammonium-15N-chloride | Sigma | 299251 | |
| 1,3 13C2 glycerol | Sigma | 492639 | |
| 2 13C glycerol | Sigma | 489484 | |
| Kanamycin | Sigma | K1876 | |
| Carbenicillin | Sigma | C3416 | |
| Sodium phosphate dibasic | Sigma | S7907 | |
| Potassium phosphate monobasic | Sigma | P5655 | |
| Sodium chloride | Sigma | 71380 | |
| calcium chloride | Sigma | C1016 | |
| Magnesium sulfate | Sigma | 208094 | |
| Iron Chloride | Sigma | 157740 | |
| Zinc chloride | Sigma | 793523 | |
| MEM Vitamin Solution (100×) | Sigma | M68954 | |
| IPTG | Fisher | BP1755 | |
| Trizma base | Sigma | T1503 | |
| Tricine | Sigma | T0377 | |
| SDS | Sigma | 436143 | |
| sodium azide | sigma | 71289 | |
| 4,4-dimethyl-4-silapentane-1-sulfonic acid | Sigma | 178837 | |
| Name | Company | Catalog Number | Comments |
| Softwares | |||
| Unicorn 6.3 | GE Healthcare | Akta systems | |
| ccpNMR | CCPN | spectrometer systems |
References
- Morag, O., Sgourakis, N. G., Baker, D., Goldbourt, A. The NMR-Rosetta capsid model of M13 bacteriophage reveals a quadrupled hydrophobic packing epitope. Proc Natl Acad Sci U S A. 112 (4), 971-976 (2015).
- Loquet, A., et al. Atomic model of the type III secretion system needle. Nature. 486 (7402), 276-279 (2012).
- Demers, J. P., et al. High-resolution structure of the Shigella type-III secretion needle by solid-state NMR and cryo-electron microscopy. Nat Commun. 5 (4976), (2014).
- Habenstein, B., et al. Hybrid Structure of the Type 1 Pilus of Uropathogenic Escherichia coli. Angew Chem Int Ed Engl. 54 (40), 11691-11695 (2015).
- Cady, S. D., et al. Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature. 463 (7281), 689-692 (2010).
- Park, S. H., et al. Structure of the chemokine receptor CXCR1 in phospholipid bilayers. Nature. 491 (7426), 779-783 (2012).
- Kaplan, M., et al. Probing a cell-embedded megadalton protein complex by DNP-supported solid-state NMR. Nat Methods. 12 (7), 649-652 (2015).
- Wang, S., et al. Solid-state NMR spectroscopy structure determination of a lipid-embedded heptahelical membrane protein. Nat Methods. 10 (10), 1007-1012 (2013).
- Daskalov, A., et al. Signal transduction by a fungal NOD-like receptor based on propagation of a prion amyloid fold. PLoS Biol. 13 (2), e1002059 (2015).
- Daskalov, A., et al. Identification of a novel cell death-inducing domain reveals that fungal amyloid-controlled cell death is related to necroptosis. Proc Natl Acad Sci U S A. , (2016).
- Li, J., et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell. 150 (2), 339-350 (2012).
- Knowles, T. P., Vendruscolo, M., Dobson, C. M. The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol. 15 (6), 384-396 (2014).
- Aguzzi, A., Lakkaraju, A. K. Cell Biology of Prions and Prionoids: A Status Report. Trends Cell Biol. 26 (1), 40-51 (2016).
- Habenstein, B., Loquet, A. Solid-state NMR: An emerging technique in structural biology of self-assemblies. Biophys Chem. , (2015).
- Meier, B. H., Bockmann, A. The structure of fibrils from ‘misfolded’ proteins. Curr Opin Struct Biol. 30, 43-49 (2015).
- Miao, Y., Cross, T. A. Solid state NMR and protein-protein interactions in membranes. Curr Opin Struct Biol. 23 (6), 919-928 (2013).
- Tang, M., Comellas, G., Rienstra, C. M. Advanced solid-state NMR approaches for structure determination of membrane proteins and amyloid fibrils. Acc Chem Res. 46 (9), 2080-2088 (2013).
- Weingarth, M., Baldus, M. Solid-state NMR-based approaches for supramolecular structure elucidation. Acc Chem Res. 46 (9), 2037-2046 (2013).
- Loquet, A., Habenstein, B., Lange, A. Structural investigations of molecular machines by solid-state NMR. Acc Chem Res. 46 (9), 2070-2079 (2013).
- Yan, S., Suiter, C. L., Hou, G., Zhang, H., Polenova, T. Probing structure and dynamics of protein assemblies by magic angle spinning NMR spectroscopy. Acc Chem Res. 46 (9), 2047-2058 (2013).
- Tycko, R., Wickner, R. B. Molecular structures of amyloid and prion fibrils: consensus versus controversy. Acc Chem Res. 46 (7), 1487-1496 (2013).
- Hong, M., Zhang, Y., Hu, F. Membrane protein structure and dynamics from NMR spectroscopy. Annu Rev Phys Chem. 63, 1-24 (2012).
- Jelinek, R., Ramamoorthy, A., Opella, S. J. High-Resolution Three-Dimensional Solid-state NMR Spectroscopy of a Uniformly 15N-Labeled Protein. J Am Chem Soc. 117, 12348-12349 (1995).
- Xu, J., et al. Bicelle-enabled structural studies on a membrane-associated cytochrome B5 by solid-state MAS NMR spectroscopy. Angew Chem Int Ed Engl. 47 (41), 7864-7867 (2008).
- Durr, U. H., Gildenberg, M., Ramamoorthy, A. The magic of bicelles lights up membrane protein structure. Chem Rev. 112 (11), 6054-6074 (2012).
- Yamamoto, K., et al. Probing the transmembrane structure and topology of microsomal cytochrome-p450 by solid-state NMR on temperature-resistant bicelles. Sci Rep. 3, 2556 (2013).
- Huang, R., et al. Probing the transmembrane structure and dynamics of microsomal NADPH-cytochrome P450 oxidoreductase by solid-state NMR. Biophys J. 106 (10), 2126-2133 (2014).
- Durr, U. H., Yamamoto, K., Im, S. C., Waskell, L., Ramamoorthy, A. Solid-state NMR reveals structural and dynamical properties of a membrane-anchored electron-carrier protein, cytochrome b5. J Am Chem Soc. 129 (21), 6670-6671 (2007).
- Hong, M. Determination of multiple phi-torsion angles in proteins by selective and extensive (13)C labeling and two-dimensional solid-state NMR. J Magn Reson. 139 (2), 389-401 (1999).
- Lundstrom, P., et al. Fractional 13C enrichment of isolated carbons using [1-13C]- or [2- 13C]-glucose facilitates the accurate measurement of dynamics at backbone Calpha and side-chain methyl positions in proteins. J Biomol NMR. 38 (3), 199-212 (2007).
- Loquet, A., Lv, G., Giller, K., Becker, S., Lange, A. 13C spin dilution for simplified and complete solid-state NMR resonance assignment of insoluble biological assemblies. J Am Chem Soc. 133 (13), 4722-4725 (2011).
- Castellani, F., et al. Structure of a protein determined by solid-state magic-angle-spinning NMR spectroscopy. Nature. 420 (6911), 98-102 (2002).
- Higman, V. A., et al. Assigning large proteins in the solid state: a MAS NMR resonance assignment strategy using selectively and extensively 13C-labelled proteins. J Biomol NMR. 44 (4), 245-260 (2009).
- Bockmann, A., et al. Characterization of different water pools in solid-state NMR protein samples. J Biomol NMR. 45 (3), 319-327 (2009).
- Cavanagh, J., Fairbrother, W. J., Palmer, A. G., Skelton, N. J. . Protein NMR spectroscopy, principles and practice. , (1996).
- Hartman, S. R., Hahn, E. L. Nuclear Double Resonance in the Rotating Frame. Phys Rev. 128 (5), 2042-2053 (1962).
- Harris, R. K., et al. Further conventions for NMR shielding and chemical shifts IUPAC recommendations 2008. Solid State Nucl Magn Reson. 33 (3), 41-56 (2008).
- Wang, Y., Jardetzky, O. Probability-based protein secondary structure identification using combined NMR chemical-shift data. Protein Sci. 11 (4), 852-861 (2002).
- Shaka, A. J., Baker, P. B., Freeman, R. Computer-Optimized Scheme for Wideband Applications and Low-Level Operation. J Magn Reson. 64, 547-552 (1985).
- Szeverenyi, N. M., Sullivan, M. J., Maciel, G. E. Observation of Spin Exchange by Two-Dimensional Fourier-Transform C-13 Cross Polarization-Magic-Angle Spinning. J Magn Reson. 47, 462-475 (1982).
- Baldus, M., Petkova, A. T., Herzfeld, J., Griffin, R. G. Cross polarization in the tilted frame: assignment and spectral simplification in heteronuclear spin systems. Mol Phys. 95 (5), 1197-1207 (1998).
- Verel, R., Ernst, M., Meier, B. H. Adiabatic dipolar recoupling in solid-state NMR: the DREAM scheme. J Magn Reson. 150 (1), 81-99 (2001).
- . Sparky – NMR Assignment and Integration Software Available from: https://www.cgl.ucsf.edu/home/sparky/ (2017)
- Luca, S., et al. Secondary chemical shifts in immobilized peptides and proteins: a qualitative basis for structure refinement under magic angle spinning. J Biomol NMR. 20 (4), 325-331 (2001).
- Shen, Y., Bax, A. SPARTA+: a modest improvement in empirical NMR chemical shift prediction by means of an artificial neural network. J Biomol NMR. 48 (1), 13-22 (2010).
- Berjanskii, M. V., Neal, S., Wishart, D. S. PREDITOR: a web server for predicting protein torsion angle restraints. Nucleic Acids Res. 34 (Web Server issue), W63-W69 (2006).
- Bardiaux, B., Malliavin, T., Nilges, M. ARIA for solution and solid-state NMR. Methods Mol Biol. 831, 453-483 (2012).
- Guerry, P., Herrmann, T. Comprehensive automation for NMR structure determination of proteins. Methods Mol Biol. 831, 429-451 (2012).
- Vasa, S., et al. beta-Helical architecture of cytoskeletal bactofilin filaments revealed by solid-state NMR. Proc Natl Acad Sci U S A. 112 (2), E127-E136 (2015).
- He, L., et al. Structure determination of helical filaments by solid-state NMR spectroscopy. Proc Natl Acad Sci U S A. 113 (3), E272-E281 (2016).
- Tang, M., et al. High-resolution membrane protein structure by joint calculations with solid-state NMR and X-ray experimental data. J Biomol NMR. 51 (3), 227-233 (2011).
- Paravastu, A. K., Leapman, R. D., Yau, W. M., Tycko, R. Molecular structural basis for polymorphism in Alzheimer’s beta-amyloid fibrils. Proc Natl Acad Sci U S A. 105 (47), 18349-18354 (2008).
- Schutz, A. K., et al. Atomic-resolution three-dimensional structure of amyloid beta fibrils bearing the Osaka mutation. Angew Chem Int Ed Engl. 54 (1), 331-335 (2015).
- Sgourakis, N. G., Yau, W. M., Qiang, W. Modeling an in-register, parallel "iowa" abeta fibril structure using solid-state NMR data from labeled samples with rosetta. Structure. 23 (1), 216-227 (2015).
- Lewandowski, J. R., De Paepe, G., Griffin, R. G. Proton assisted insensitive nuclei cross polarization. J Am Chem Soc. 129 (4), 728-729 (2007).
- Carlon, A., et al. How to tackle protein structural data from solution and solid state: An integrated approach. Prog Nucl Magn Reson Spectrosc. 92-93, 54-70 (2016).
- Judge, P. J., Taylor, G. F., Dannatt, H. R., Watts, A. Solid-state nuclear magnetic resonance spectroscopy for membrane protein structure determination. Methods Mol Biol. 1261, 331-347 (2015).
- Wang, S., Ladizhansky, V. Recent advances in magic angle spinning solid state NMR of membrane proteins. Prog Nucl Magn Reson Spectrosc. 82, 1-26 (2014).
- Sborgi, L., et al. Structure and assembly of the mouse ASC inflammasome by combined NMR spectroscopy and cryo-electron microscopy. Proc Natl Acad Sci U S A. 112 (43), 13237-13242 (2015).
- Loquet, A., et al. Atomic structure and handedness of the building block of a biological assembly. J Am Chem Soc. 135 (51), 19135-19138 (2013).
- Walti, M. A., et al. Atomic-resolution structure of a disease-relevant Abeta(1-42) amyloid fibril. Proc Natl Acad Sci U S A. 113 (34), E4976-E4984 (2016).
- Colvin, M. T., et al. Atomic Resolution Structure of Monomorphic Abeta42 Amyloid Fibrils. J Am Chem Soc. 138 (30), 9663-9674 (2016).
- Xiao, Y., et al. Abeta(1-42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer’s disease. Nat Struct Mol Biol. 22 (6), 499-505 (2015).
- Tuttle, M. D., et al. Solid-state NMR structure of a pathogenic fibril of full-length human alpha-synuclein. Nat Struct Mol Biol. 23 (5), 409-415 (2016).
- Wasmer, C., et al. Amyloid fibrils of the HET-s(218-289) prion form a beta solenoid with a triangular hydrophobic core. Science. 319 (5869), 1523-1526 (2008).
- Van Melckebeke, H., et al. Atomic-resolution three-dimensional structure of HET-s(218-289) amyloid fibrils by solid-state NMR spectroscopy. J Am Chem Soc. 132 (39), 13765-13775 (2010).
- Lamley, J. M., et al. Solid-state NMR of a protein in a precipitated complex with a full-length antibody. J Am Chem Soc. 136 (48), 16800-16806 (2014).
- Agarwal, V., et al. De novo 3D structure determination from sub-milligram protein samples by solid-state 100 kHz MAS NMR spectroscopy. Angew Chem Int Ed Engl. 53 (45), 12253-12256 (2014).
- Stanek, J., et al. NMR Spectroscopic Assignment of Backbone and Side-Chain Protons in Fully Protonated Proteins: Microcrystals, Sedimented Assemblies, and Amyloid Fibrils. Angew Chem Int Ed Engl. 55 (50), 15504-15509 (2016).
- Baker, L. A., Baldus, M. Characterization of membrane protein function by solid-state NMR spectroscopy. Curr Opin Struct Biol. 27, 48-55 (2014).
- Luchinat, E., Banci, L. In-cell NMR: a topical review. IUCrJ. 4 (Pt 2), 108-118 (2017).
- Freedberg, D. I., Selenko, P. Live cell NMR. Annu Rev Biophys. 43, 171-192 (2014).
- Selenko, P., Wagner, G. Looking into live cells with in-cell NMR spectroscopy. J Struct Biol. 158 (2), 244-253 (2007).
- Qiang, W., Yau, W. M., Luo, Y., Mattson, M. P., Tycko, R. Antiparallel beta-sheet architecture in Iowa-mutant beta-amyloid fibrils. Proc Natl Acad Sci U S A. 109 (12), 4443-4448 (2012).
- Bateman, D. A., Tycko, R., Wickner, R. B. Experimentally derived structural constraints for amyloid fibrils of wild-type transthyretin. Biophys J. 101 (10), 2485-2492 (2011).
- Marchanka, A., Simon, B., Althoff-Ospelt, G., Carlomagno, T. RNA structure determination by solid-state NMR spectroscopy. Nat Commun. 6, 7024 (2015).
- Schutz, A. K., et al. The amyloid-Congo red interface at atomic resolution. Angew Chem Int Ed Engl. 50 (26), 5956-5960 (2011).
