This protocol provides a step-wise description of how human iPSCs can be transferred from feeder layer to feeder-free conditions, and subsequently propagated in a limited manner to specifically enable cost-effective immunocytochemistry for confirming pluripotency maintenance. Figure 1 shows a schematic representation of this protocol. Figure 2A shows hiPSC colonies growing on iMEFs in 6-well plates. These colonies exhibit typical morphology with defined borders and dense phase bright centers. As shown in Figure 2B, after the transfer of iPSCs to feeder-free conditions in 12-well plates, colony morphology appears somewhat chaotic with less distinct edges. Also, some iMEFs may remain in the culture at this stage. Figure 2C shows that after an additional passage in the feeder-free system, iPSC colonies display a classic monolayer morphology with a high nucleus to cytoplasm ratio. At this stage, it is seen that iMEFs have been virtually eliminated from the culture.
Colonies growing under feeder-free conditions are mechanically picked and plated onto small multi-well chamber slides or glass coverslips, and probed for specific pluripotency antigens via immunocytochemistry. Figure 3 shows representative colonies that are immunopositive for SSEA4 or Tra-1-60 (3B, 3F, surface pluripotency antigens) and Oct-4 or Sox2 (3 A, 3E, intracellular pluripotency antigens).
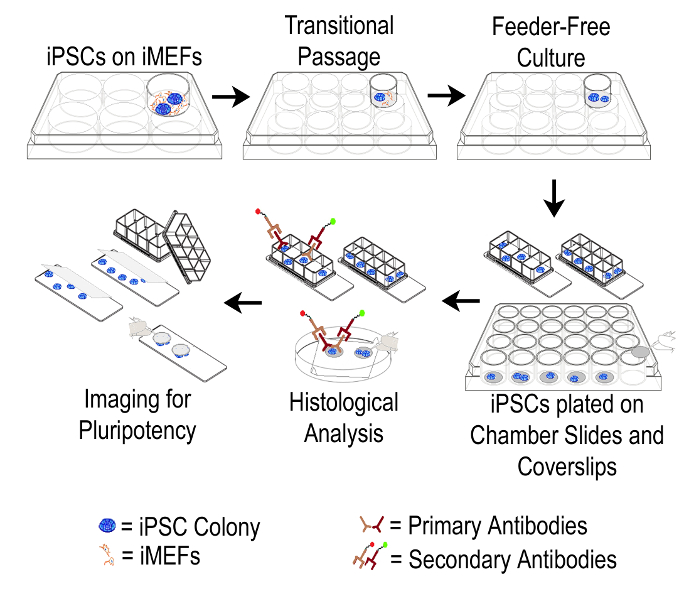
Figure 1: Schematic Representation of the Protocol. Overall schema of the described method used to transition human iPSCs from iMEF feeders to feeder-free culture and subsequent probing of cells with pluripotency markers. iMEF: irradiated Mouse Embryonic Fibroblast; iPSC: Induced Pluripotent Stem Cell. Please click here to view a larger version of this figure.
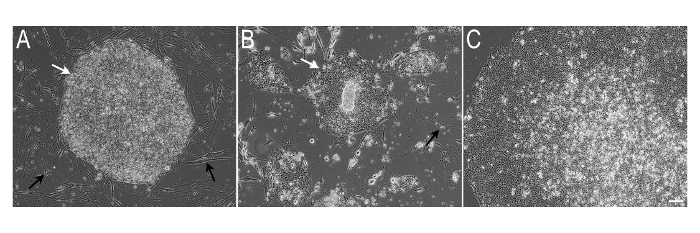
Figure 2. Representative Images of iPSC Colonies Transitioning from iMEF Feeders to Feeder-free Culture Conditions. A) Dense iPSC colony (white arrow) grown on iMEF feeder cells (black arrow). B) After the initial passage on to a matrix-coated 12-well plate in serum-free medium, a few iMEFs may still be observed in culture (black arrow). C) Typical morphology of a monolayer iPSC colony grown feeder-free. No iMEFs remain in culture at this stage. Scale Bar (A-C) = 100 µm. Please click here to view a larger version of this figure.
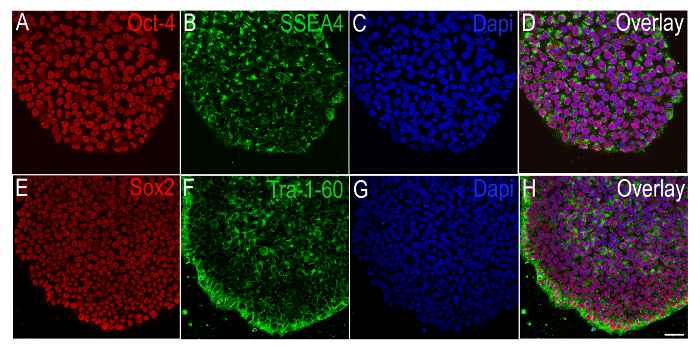
Figure 3. Immunocytochemical Characterization of Human iPSCs Plated in Small Well Plates. Representative immunofluorescence images of iPSCs, obtained via a confocal microscope, showing the positive expression of pluripotency markers A) Oct-4 (red) and B) SSEA4 (green) with C) the nuclear stain DAPI (blue), and E) Sox2 (red) and F) Tra-1-60 (green) with G) DAPI (blue). D and H show the overlay images relating to A-C and E-G. Scale bar (A-H) = 100 µm. Please click here to view a larger version of this figure.
| hiPSC Media for maintenance on Feeders | ||
| Component | Stock Concentration | Final Concentration |
| DMEM-F12/HEPES | 100% | 80% |
| Knockout Serum Replacement | 100% | 20% |
| L-Glutamine | 200 mM | 1 mM |
| MEM-NEAA | 10 mM | 0.1 mM |
| 2-mercaptoethanol | 55 mM | 0.1 mM |
| Recombinant Human FGF-Basic | 10 µg/mL | 10 ng/mL |
| Y-27632 ROCK Inhibitor | 10 µM | |
| Matrigel coating for Feeder-Free cultures | ||
| Component | Amount | |
| Matrigel hESC-qualified Matrix | Dilution Factor 269 µL | |
| DMEM-F12/HEPES | 25 mL | |
| Note: Dilution Factor is lot dependent and must be ascertained from certificate of analysis | ||
| mTeSR1 complete media for Feeder-Free cultures | ||
| Component | Amount | |
| mTeSR1 Basal Medium | 400 mL | |
| mTeSR1 5x Supplement | 100 mL | |
| Note: Once mixed, mTeSR1 complete media may be frozen in aliquots and used until component expiration date | ||
| Note: mTeSR1 complete media must be warmed at room temperature only. Do not place in water bath | ||
Table 1: iPSC Culture Media Recipes and Plate Coatings.
| 4% Paraformaldehyde fixative | |
| Component | Amount |
| 0.1 M PO4 Buffer | 2 L |
| Paraformaldehyde, prill | 80 g |
| ICC Blocking Solution without Triton-X-100 | |
| Component | Amount |
| 1x Phosphate Buffered Saline | 49 mL |
| Normal Goat Serum | 1 mL |
| Bovine Serum Albumin | 0.5 g |
| ICC Blocking Solution with Triton-X-100 | |
| Component | Amount |
| 1x Phosphate Buffered Saline | 49 mL |
| Normal Goat Serum | 1 mL |
| Bovine Serum Albumin | 0.5 g |
| Triton-X-100 | 200 µL |
Table 2: Fixative and Immunocytochemistry Recipes.
