When the leptomeninges processing protocol has been successful, outgrowth of meningeal fibroblasts is first observed three to nine days after dissection, although this can depend on the length of post-mortem interval for the brain. Figure 1 demonstrates meningeal fibroblast cultures of four different donors. Figure 1A shows a leptomeninges piece held down by a glass cover slip (dark diagonal line) and fibroblast outgrowth around the tissue four days after processing from an 88-year-old donor with 10 h 20 min post-mortem interval. Figure 1B shows sparse fibroblast outgrowth seven days post processing from a 70-year-old donor and 11 h 45 min post-mortem interval. Figure 1C shows fibroblast outgrowth 13 days after dissection from an 88-year-old donor and 24 h post-mortem interval. Figure 1D shows a confluent culture that was passaged into a T75 vessel and cells can be cryopreserved at this stage.
Meningeal fibroblasts are bipolar or multipolar and have elongated or irregular shapes (Figure 2). They are characterized by the expression of glycoprotein fibronectin and fibroblast marker SERPINH which is localized to the endoplasmic reticulum (Figures 2A-2C). Meningeal fibroblasts are not immuno-reactive to transcription factor SRY (Sex Determining Region Y)-Box 2 (SOX2) which is a marker for undifferentiated stem cells (Figure 2A). However, a subset of leptomeningeal fibroblasts is positive for type VI intermediate filament nestin, a neural stem cell marker (Figure 2B), and neuron-specific class III beta-tubulin (TUJ1) (Figure 2C).
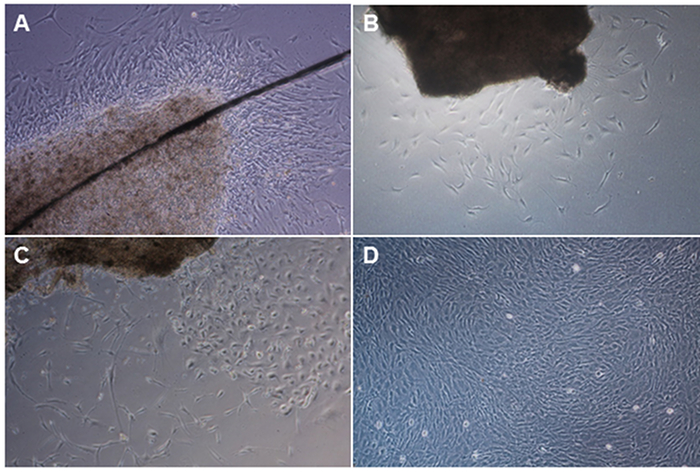
Figure 1. Examples of outgrowth of meningeal fibroblasts from four different donors. A) Leptomeninges tissue piece is held down by a glass cover slip (dark diagonal line) and fibroblast outgrowth around the tissue was observed four days after processing from an 88-year-old donor with 10 h 20 min post-mortem interval. B) Sparse fibroblast outgrowth seven days post processing from a 70-year-old donor and 11 h 45 min post-mortem interval. C) Meningeal fibroblast outgrowth 13 days after dissection from an 88-year-old donor and 24 h post-mortem interval. D) Confluent meningeal fibroblast culture passaged into a T75 vessel. Please click here to view a larger version of this figure.
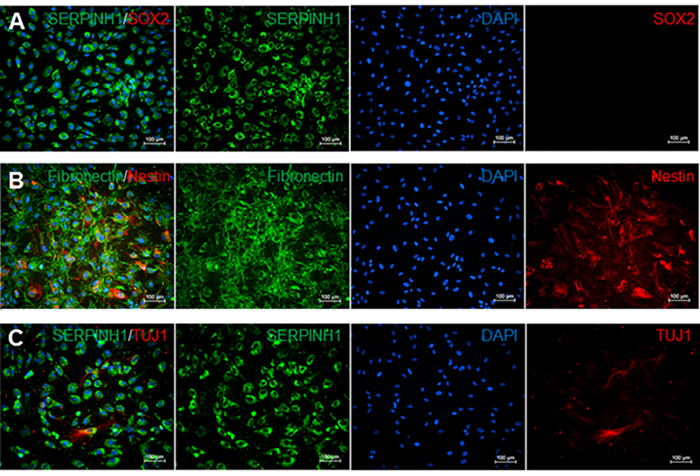
Figure 2. Immunostaining for meninges, stem cell, neural stem cell, and neuron markers. A) Immunofluorescence staining for fibroblast markers SERPINH1 (green) and stem cell marker SOX2 (red). SERPINH1 was expressed in all counted cell nuclei. Stem cell marker SOX2 was not detected in these leptomeninges culture. B) Immunofluorescence staining for meninges-specific marker fibronectin (green) and neural stem cell marker NESTIN (red). 52.1% of counted cell nuclei were positive for both NESTIN and Fibronectin. C) Immunofluorescence staining for SERPINH1 (green) and neuronal marker TUJ1 (red). 6.9% of counted nuclei were positive for SERPINH1 and TUJ1. Cell count data were determined by using the manual Cell Counter Multi-Select tool on ImageJ. Single stained images were assessed by counting the number of DAPI-positive cells compared to the number of cells positive for each marker in percentages. Please click here to view a larger version of this figure.
| Day | Expected results | |||||
| 3-9 | Outgrowth of first meningeal fibroblasts. | |||||
| 7-18 | Meningeal fibroblasts expand, and culture becomes denser. Media change of 2 mL every other day. | |||||
| 18-25 | 6-well plate becomes confluent, once meningeal fibroblasts are confluent combine three wells into a T75 culture flask (passage at 1:3 to 1:4). | |||||
| 26-45 | One confluent T75 culture flask results in 5-7 million cells. Cryopreserve meningeal fibroblasts at 1 million cells/vial. | |||||
Table 1: Timeline of cellular outgrowth.
