Identification of Host Pathways Targeted by Bacterial Effector Proteins using Yeast Toxicity and Suppressor Screens
Summary
Bacterial pathogens secrete proteins into the host that target crucial biological processes. Identifying the host pathways targeted by bacterial effector proteins is key to addressing molecular pathogenesis. Here, a method using a modified yeast suppressor and toxicity screen to elucidate host pathways targeted by toxic bacterial effector proteins is described.
Abstract
Intracellular bacteria secrete virulence factors called effector proteins into the host cytosol that act to subvert host proteins and/or their associated biological pathways to the benefit of the bacterium. Identification of putative bacterial effector proteins has become more manageable due to advances in bacterial genome sequencing and the advent of algorithms that allow in silico identification of genes encoding secretion candidates and/or eukaryotic-like domains. However, identification of these important virulence factors is only an initial step. Naturally, the goal is to determine the molecular function of effector proteins and elucidate how they interact with the host. In recent years, techniques like the yeast two-hybrid screen and large-scale immunoprecipitations coupled with mass spectrometry have aided in the identification of protein-protein interactions. Although identification of a host binding partner is the crucial first step toward elucidating the molecular function of a bacterial effector protein, sometimes the host protein is found to have multiple biological functions (e.g., actin, clathrin, tubulin), or the bacterial protein may not physically bind host proteins, depriving the researcher of crucial information about the precise host pathway being manipulated. A modified yeast toxicity screen coupled with a suppressor screen has been adapted to identify host pathways impacted by bacterial effector proteins. The toxicity screen relies on a toxic effect in yeast caused by the effector protein interfering with the host biological pathways, which often manifests as a growth defect. Expression of a yeast genomic library is used to identify host factors that suppress the toxicity of the bacterial effector protein and thus identify proteins in the pathway that the effector protein targets. This protocol contains detailed instructions for both the toxicity and suppressor screens. These techniques can be performed in any lab capable of molecular cloning and cultivation of yeast and Escherichia coli.
Introduction
The first report of procedures similar to those presented here characterized the Legionella pneumophila type IV effector SidD, a deAMPylase that modifies Rab11. Comparable techniques were used for the characterization of several L. pneumophila effectors1,2,3. The assay was adapted to characterize a Coxiella burnetii type IV effector protein4, and recently the utility of this technique was expanded for the characterization of Chlamydia trachomatis inclusion membrane proteins5.
This protocol can be broken into two major parts: 1) the yeast toxicity screen, in which the bacterial effector protein of interest is expressed in yeast and clones are screened for a toxic phenotype as evidenced by a growth defect, and 2) the yeast suppressor screen, in which the toxic phenotype is suppressed by expression of a yeast genomic library in the toxic strain. Thus, the toxicity screen is a screen for toxic phenotypes that manifest as growth defects when the bacterial effector of interest is overexpressed. Toxic clones, successfully transformed with and expressing the bacterial effector, are selected and saved for the next step. The second major step involves overexpressing a partially digested yeast genomic library in the toxic yeast clone. Plasmids making up the yeast genomic library suggested for the use in this protocol carry 5−20 kb inserts, usually corresponding to 3−13 yeast open reading frames (ORF) of an average gene size of ~1.5 kb across all plasmids, representing the whole yeast genome covered approximately 10x. This part of the assay is called the suppressor screen, as the goal is to suppress the toxicity of the bacterial effector protein. Potential suppressor plasmids are isolated from yeast, sequenced, and the suppressing ORFs identified. The rationale underlying the suppressor screen is that the effector protein binds, interacts with, and/or overwhelms components of the host pathway it targets, and that providing those host proteins back in excess can rescue the toxic effect on the pathway and thus, the growth defect. Thus, identified ORFs that suppress toxicity often represent multiple participants of a host pathway. Orthogonal experiments are then performed to verify that the bacterial effector indeed interacts with the implicated pathway. This is especially necessary if a binding partner such as clathrin or actin has been identified, because these proteins are involved in a multitude of host processes. Further experiments can then elucidate the physiological function of the effector protein during infection. The toxicity and suppressor screens are also powerful tools for deciphering the physiological function of bacterial effector proteins that do not physically bind host proteins with affinities sufficient to detect by immunoprecipitation or that interact with the host in enzymatic hit-and-run interactions that may not be detected by a yeast-two hybrid screen.
Although the suppressor screen can be a powerful method to reveal potential physiological interactions between bacterial effector proteins and host pathways, the bacterial effector protein must induce a growth defect in yeast, otherwise using it in the suppressor screen will be of little use. Furthermore, the toxic phenotype must result in at least a 2−3 log10 deficit in growth or it will be difficult to identify suppressors. If a laboratory is set up for cell culture, screening effector proteins for toxicity in common cell lines such as HeLa can often give insight as to whether it is worth the effort to proceed with the yeast toxicity screen. Ectopic expression of the effector protein in HeLa cells sometimes results in toxicity that correlates very strongly with toxicity in the yeast strain used for these screens4. Observable hallmarks of stress in HeLa cells include loss of stress fibers, cell detachment from the plate, and nuclear condensation indicating apoptosis. Any visual indication of stress in HeLa cells make the protein of interest a good candidate for inducing a growth defect in yeast, which replicate much more rapidly and are thus more responsive to perturbation of essential pathways.
It should be noted that the suppressor screen does not always identify host binding partners as suppressors, but it can still implicate critical components of the host pathway(s) targeted, yielding a holistic view of the biological processes being hijacked by the bacterial effector protein. On the surface, this seems counterintuitive, because providing the binding partner of the effector protein in excess would be expected to rescue the growth defect. In efforts to identify pathways targeted by the C. trachomatis effector protein CT229 (CpoS), which binds to at least 10 different Rab GTPases during infection5, none of the Rab binding partners suppressed the toxicity of CT229. However, numerous suppressors involved in clathrin-coated vesicle (CCV) trafficking were identified, which led to further work demonstrating that CT229 specifically subverts Rab-dependent CCV trafficking. Similarly, when investigating the C. burnetii effector protein Cbu0041 (CirA) several Rho GTPases that rescued the yeast growth defect were identified, and it was later found that CirA functions as a GTPase activating protein (GAP) for RhoA4.
The usefulness of the yeast suppressor screen for elucidating host pathways targeted by bacterial effector proteins cannot be overstated, and other researchers attempting to characterize intracellular bacterial effector proteins can greatly benefit from these techniques. These assays are of value if immunoprecipitations and/or yeast-two hybrid screens have failed to find a binding partner and can elucidate which pathways are targeted by the bacterial effector protein. Here, detailed protocols for the toxicity and suppressor screens to identify host biological pathways targeted by intracellular bacterial effector proteins are provided, as well as some of the common obstacles experienced when using these assays and their corresponding solutions.
Protocol
1. Preparation of media and reagents
NOTE: Plates should be prepared before the day of the assay and are good for 1 month. Media and reagents can be made at any point and are good for 1 month.
- Prepare 1 L of the glucose solution (10% w/v) by dissolving 100 g of D-(+)-glucose in 800 mL of distilled water in a 1, 000 mL beaker. Adjust the volume to 1 L with distilled water. Filter through a 0.2 µm sterile filter into a sterile 1 L media storage bottle.
- Prepare 1 L of the galactose solution (10% w/v) by dissolving 100 g of D-(+)- galactose in 800 mL of distilled water in a 1 L beaker. Adjust the volume to 1 mL using distilled water and filter through a 0.2 µm sterile filter into a sterile 1,000 mL media storage bottle.
- Prepare 1 L of yeast extract peptone dextrose (YPD) agar by dissolving 10 g of yeast extract, 20 g of peptone, 20 g of glucose, and 20 g of agar in 1,000 mL of distilled water. Autoclave for 20 min and cool in 56 ˚C water bath until cooled. Pour into 100 mm plates using ~20 mL of media per plate.
- Prepare 1 L of YPD broth by dissolving 10 g of yeast extract, 20 g of peptone, and 20 g of glucose in 1,000 mL of distilled water. Autoclave for 20 min.
- Prepare the synthetic dropout (SD) uracil (Ura–) glucose agar. For 1 L, dissolve 6.7 g of yeast nitrogen base without amino acids, 1.9 g of dropout supplement without uracil, and 15 g of agar in 800 mL of distilled water. Autoclave for 20 min and cool in a 56 ˚C water bath until the temperature is ~50−60 ˚C. Using a 50 mL serological pipette or a sterile graduated cylinder, add 200 mL of the glucose solution prepared in step 1.1. Mix well by gently swirling or place on stir plate. Pour into 100 mm plates using ~20 mL of media per plate.
- Prepare the synthetic dropout (SD) uracil (Ura–) galactose agar. For 1 L, dissolve 6.7 g of yeast nitrogen base without amino acids, 1.9 g of dropout supplement without uracil, and 15 g of agar in 800 mL of distilled water. Autoclave for 20 min and cool in a 56 ˚C water bath until the temperature is ~50−60 ˚C. Using a 50 mL serological pipette or a sterile graduated cylinder, add 200 mL of the galactose solution prepared in step 1.2. Mix well by gently swirling or place on the stir plate. Pour into 100 mm plates using ~20 mL of media per plate.
- Prepare the synthetic dropout (SD) uracil (Ura–) glucose broth. For 1 L, dissolve 6.7 g of yeast nitrogen base without amino acids and 1.9 g of dropout supplement without uracil in 800 mL of distilled water. Autoclave for 20 min and cool in a 56 ˚C water bath until the temperature is ~50−60 ˚C. Using a 50 mL serological pipette or a sterile graduated cylinder, add 200 ml of the glucose solution prepared in step 1.1.
- Prepare the synthetic dropout (SD) uracil (Ura–) leucine (Leu–) glucose agar. For 1 L, dissolve 6.7 g of the yeast nitrogen base without amino acids; 1.9 g of dropout supplement without uracil, leucine, and tryptophan; 15 g of agar; and 0.076 g of tryptophan in 800 mL of distilled water. Autoclave for 20 min and cool in a 56 ˚C water bath until the temperature is ~50−60 ˚C. Using a 50 mL serological pipette or a sterile graduated cylinder, add 200 mL of the glucose solution prepared in step 1.1. Pour into 100 mm plates using ~20 mL of media per plate.
- Prepare synthetic dropout (SD) uracil (Ura–) leucine (Leu–) galactose agar. For 1 L, dissolve 6.7 g of yeast nitrogen base without amino acids; 1.9 g of dropout supplement without uracil, leucine, and tryptophan; 15 g of agar; and 0.076 g of tryptophan in 800 mL of distilled water. Autoclave for 20 min and cool in a 56 ˚C water bath until the temperature is ~50−60 ˚C. Using a 50 mL serological pipette or a sterile graduated cylinder, add 200 mL of the galactose solution prepared in step 1.2. Mix well by gently swirling or placing on a stir plate. Pour into 100 mm plates using ~20 mL of media per plate.
- Prepare synthetic dropout (SD) uracil (Ura–) leucine (Leu–) glucose broth. For 1 L, dissolve 6.7 g of yeast nitrogen base without amino acids; 1.9 g of dropout supplement without uracil, leucine, tryptophan; and 0.076 g of tryptophan in 800 mL of distilled water. Autoclave for 20 min and cool in a 56−60 ˚C water bath until the temperature is ~50 ˚C. Using a 50 mL serological pipette or a sterile graduated cylinder, add 200 mL of the glucose solution prepared in step 1.1.
- Prepare polyethylene glycol (PEG) solution. Add 50% w/v of polyethylene glycol 3350 in distilled water. Sterilize by autoclaving.
- Prepare 1 M lithium acetate (LiAc) by dissolving 10.2 g of lithium acetate dehydrate in 200 mL of distilled water. Sterilize by autoclaving.
- Prepare herring sperm DNA. Dilute 10 mg/mL herring sperm DNA to 2 mg/mL using distilled water. Heat at 100 ˚C for 5 min and immediately place on ice for 5 min.
2. Cloning the gene of interest into the yeast toxicity plasmid pYesNTA-Kan
NOTE: Currently, there are a variety yeast toxicity vectors available both commercially and academically. The yeast suppressor screen can be used in conjunction with many of these vectors provided the plasmid expressing the effector protein of interest uses a dropout selection other than uracil and an antibiotic marker other than BlaR. This work used a modified pYesNTA vector4,5 that includes a kanamycin resistance cassette for easier screening for potential suppressors. The cloning scheme must allow for the effector protein to be in-frame with the Gal promoter and His-Tag. The Chlamydia trachomatis inclusion membrane protein CT229 was used as a proof of principle for these assays.
- Use PCR to amplify CT229 from C. trachomatis genomic DNA following the manufacturer's instructions using CT229 +1 Kpn F (CCGGTACCAATGAGCTGTTCTAATGTTAATTCAGGT) and CT229 XhoI R (CCCTCGAGTTTTTTACGACGGGATGCC) primers. Use the following PCR conditions: (1) 98 °C for 30 s; (2) 98 °C for 10 s, 55 °C for 30 s, 72 °C for 2 min; (3) repeat step 2 for a total of 25x; (4) 72 °C for 10 min; and (5) 4 °C hold.
- Analyze 5 µL of the PCR product on a 1% agarose gel (Figure 1).
- Purify the remaining 45 µL of DNA using a PCR purification kit following the manufacturer's instructions.
- Digest the 50 µL of purified PCR insert and pYesNTA-Kan (Figure 2) for 1 h at 37 °C in a water bath using KpnI-HF and XhoI-HF.
NOTE: For pYesNTA-Kan, digest 5 µg of plasmid using 5 µL of KpnI-HF, 5 µl of XhoI-HF, and 6 µL of buffer in a total volume of 60 µL. For the insert, digest the entire 50 µL of purified PCR product using 2 µL of KpnI-HF, 2 µL of XhoI-HF, and 6 µL of buffer. - Run the entire plasmid digest on a 1% agarose gel. Perform purification of the digested plasmid using a gel extraction kit following the manufacturer's instructions.
- Purify the digested PCR insert using a PCR purification kit following the manufacturer's instructions.
- Clone the insert into pYesNTA-Kan using 2 µL of pYesNTA-Kan (step 2.5), 2 µL of DNA ligase buffer, 1 µL of T4 ligase, and 15 µL of insert (step 2.6). Incubate at room temperature for 1 h.
- Add the entire cloning reaction to 50 µL of E. coli competent cells and perform the transformation reaction.
- Plate the entire transformation reaction on an LB plate containing 100 µg/mL of carbenicillin.
- Inoculate 10 mL of the LB containing 100 µg/mL of carbenicillin with three individual colonies. Incubate overnight at 37 ˚C with shaking at 150 rpm.
- Isolate the plasmid using a plasmid miniprep kit following the manufacturer's instructions.
- Sequence the isolated plasmids using gene-specific primers (step 2.1).
3. Test the protein of interest for toxicity in yeast
- Streak Saccharomyces cerevisiae W303 on YPD agar (step 1.3) to obtain isolated colonies. Incubate at 30 °C for 24 h.
- Inoculate 10 mL of YPD broth (step 1.4) with a single colony from the agar plate (step 3.1). Incubate at 30 °C with shaking at 150 rpm overnight.
- Add 0.5 mL of the overnight culture to 10 mL of YPD broth (step 1.4) and incubate at 30 °C with shaking at 150 rpm for 4 h.
- Pellet the 10 mL of culture at 3,000 x g for 10 min at 4 °C.
- Resuspend the pellet in 1 mL of sterile water, transfer to the microcentrifuge tube, and pellet at 3,000 x g for 1 min at room temperature.
- Resuspend the pellet in 1 mL of 1 mM lithium acetate (LiAc) and pellet at 3,000 x g for 1 min at room temperature.
- Repeat the LiAc wash 2x.
- Remove the wash. Resuspend the pellet in 2.4 mL of 50% PEG 3350. Add 360 µL of 1M LiAc, 500 µL of 2 mg/mL of herring sperm DNA, and 400 µL of sterile water.
NOTE: This is sufficient for 20 transformations. - Add 180 µL of the transformation mix from step 3.3.5 to a microcentrifuge tube containing 5 µL of pYesNTA-Kan CT229 plasmid DNA (100−500 ng) from step 2.12.
- Incubate in a water bath at 30 °C for 30 min.
- Incubate in a water bath at 42 °C for 30 min.
- Pellet at 3,000 x g for 1 min at room temperature.
- Remove the transformation mix with the pipette. Resuspend the pellet in 100 µL of sterile water. Plate the transformation on SD Ura– agar with glucose (step 1.5).
- Incubate plates at 30 ˚C for 48 h.
- Inoculate 5 mL of SD Ura– broth containing glucose (step 1.7) with a single colony from the plate. Incubate overnight at 30 °C with shaking at 150 rpm. Include yeast transformed with the vector alone as a negative control.
- Add 180 µL of sterile water to 5 wells of a 96 well plate (A2−A6).
- Vortex the overnight culture to mix.
- Add 180 µL of yeast to the first well (A1). Serially dilute 1:10 (six samples in total including undiluted).
- Using a multichannel pipette, spot 5 µL of each dilution on SD Ura– glucose (step 1.5) and Ura– galactose (step 1.6) agar plates. Incubate at 30 °C for 48 h.
- Assess the toxicity by comparing the growth of the yeast expressing the effector protein of interest grown on the galactose-containing media to the growth of yeast expressing the vector alone.
- Confirm the expression of the His-tag fusion protein by Western blotting.
4. Transform toxic yeast with the yeast genomic library
- Inoculate 100 mL of SD Ura– glucose broth (step 1.7) using 1 mL of the stock from step 3.4. Incubate for 16−24 h at 30 °C with shaking at 150 rpm. Place 900 mL of SD Ura– glucose broth at 30 °C overnight to prewarm the media.
- Add the entire 100 mL of the overnight culture to the prewarmed 1 L flask. Incubate for 4−5 h at 30 °C with shaking at 150 rpm.
- Pellet the culture at 6,000 x g for 10 min at 4 °C.
- Discard the supernatant. Resuspend the pellet in 250 mL of sterile water. Pellet the culture at 6,000 x g for 5 min at 4 °C.
- Discard the supernatant. Resuspend the pellet in 250 mL of 1 mM LiAc. Pellet the culture at 6,000 x g for 5 min at 4 °C.
- Remove LiAc and resuspend the pellet in 9.6 mL of 50% PEG 3350. Add 1.44 mL of 1M LiAc, 2 mL of 2 mg/mL herring sperm DNA, 50 µg of the pYep13 genomic library (ATCC 37323). Adjust the volume to 15 mL with sterile water. Mix gently by inversion.
- Incubate in a water bath at 30 °C for 30 min.
- Add 750 µL of dimethyl sulfoxide (DMSO) to enhance the transformation efficiency. Incubate in a water bath at 42 °C for 30 min. Mix by gentle inversion every 10 min.
- Pellet the yeast at 3,000 x g for 5 min at room temperature.
- Discard the supernatant and resuspend the pellet in 10 mL of sterile water. Pellet at 3,000 x g for 5 min at room temperature.
- Resuspend the pellet in 8 mL of sterile water.
- To determine the transformation efficiency, dilute 1:10 and plate 100 µL of each dilution on SD Ura– Leu– glucose agar (step 1.8).
- Plate 200 µL of the sample on SD Ura– Leu– galactose agar (step 1.9) plates. Use 50 plates in total.
- Incubate at 30 °C for 48−96 h or until colonies appear.
NOTE: Colonies generally appear on glucose agar plates at 48−72 h and galactose agar plates at 72−96 h.
- Patch colonies (potential rescues) on SD Ura– Leu– galactose agar (step 1.9) to expand and make stock. Incubate at 30 °C for 24−48h.
- Inoculate 5 mL of SD Ura– Leu– glucose broth (step 1.10) using the part of the patch. Incubate overnight at 26 °C with shaking at 150 rpm.
- Add 180 µL of the sterile water to 5 wells of a 96 well plate (A2−A6).
- Vortex the overnight culture to mix.
- Add 180 µL of yeast to the first well (A1). Serially dilute 1:10 (six samples in total including undiluted).
- Using a multichannel pipette, spot 5 µL of each dilution on SD Ura– glucose and SD Ura– galactose agar plates. Include toxic effector alone as a control.
- Incubate at 30 °C for 48 h.
- Compare the growth of the yeast expressing the toxic effector alone to yeast containing the potential suppressors. Only proceed with potential suppressors that have diminished toxicity compared to the yeast expressing the effector alone.
5. Identify and confirm suppressors
- Inoculate 5 mL of SD Ura– Leu– glucose broth (step 1.10) with 100 µL of yeast from step 4.4 and incubate overnight at 30 °C with shaking at 150 rpm.
- Pellet the yeast at 3,000 x g for 2 min at room temperature. Gently discard the supernatant using a pipette.
- Isolate the plasmid with a yeast plasmid miniprep kit following the manufacturer's instructions.
- Transform the isolated plasmid into the E. coli competent cells and plate the entire transformation on LB agar with 100 µg/mL of carbenicillin. Incubate at 37 °C for 24 h.
- Inoculate 10 mL of LB broth containing 100 µg/mL of carbenicillin, with three colonies from the plate and incubate at 37 °C overnight with shaking at 150 rpm.
- Pellet cultures at 3,000 x g for 10 min at room temperature. Isolate plasmid using a miniprep kit following the manufacturer's instructions.
- Retransform the toxic yeast with the isolated plasmids from step 5.3.1 following the steps in part 3 of the protocol.
- Inoculate 5 mL of SD Ura– Leu– glucose broth with a colony from the transformation plate. Incubate overnight at 30 °C with shaking at 150 rpm.
- Spot on Ura– Leu– glucose and galactose agar to confirm suppression of toxicity.
- Sequence using pYep13 F (ACTACGCGATCATGGCGA) and pYep13 R (TGATGCCGGCCACGATGC) primers to identify yeast ORFs.
Representative Results
Before the actual yeast suppressor screen can be performed, the effector protein of interest must be tested for toxicity in yeast. This is accomplished by expressing the protein of interest in yeast under the control of a galactose-inducible promoter. Growth on glucose (noninducing conditions) should first be compared to ensure toxicity is specifically due to the expression of the protein of interest and is not a general defect. As shown in Figure 3, toxicity manifests as smaller colonies and/or reduced growth. A 2−3 log decrease in yeast growth is ideal for the yeast suppressor screens.
As shown in Figure 4, the toxicity and suppressor screen can be accomplished in eight steps using the methods described in the protocol section. The gene of interest is cloned into a suitable vector1, and the resulting constructs are transformed into yeast to assess toxicity. This study used CT229 as the gene of interest and pYesNTA-Kan as the vector. Additionally, the expression of the His-tagged fusion protein was confirmed by Western blotting. Ideally, a 2−3 log reduction in yeast grown on galactose-containing media should be observed. If the protein of interest is toxic to yeast (Figure 3 and Figure 4), the suppressor screen can be conducted by transforming the toxic strain with the yeast genomic library pYep13. Transformants are plated on glucose agar to determine transformation efficiency and galactose agar to identify potential suppressors. Using this protocol, a minimum transformation efficiency of 5 x 105 was achieved with a total of 10−250 colonies on galactose agar. Patching these colonies on SD Ura– Leu– galactose agar (Figure 4) aided in identification of true suppressors, because many false positives will not grow when patched. Here, 50 colonies were patched and eight clones that suppressed effector toxicity were obtained (Figure 4). Spotting suppressors on SD Ura– Leu– galactose agar was done to confirm suppression of toxicity. As shown in Figure 4, pSup1 and pSup 2 suppressed the toxicity of the effector protein, whereas pSup3 did not. Therefore, pSup3 was discarded. Plasmids were subsequently isolated from the suppressors and transformed into E. coli to increase the plasmid yield. The plasmid can then be retransformed into the toxic yeast to confirm that the isolated plasmid definitely suppresses effector toxicity (Figure 4). While most of the isolated plasmids should rescue toxicity, occasionally a plasmid that does not suppress toxicity when retransformed into the toxic yeast strain is obtained. Those plasmids are discarded, and only those that suppress toxicity following retransformation should be sequenced.
The isolated plasmid will contain multiple yeast ORFs4. To identify which is the true suppressor, each ORF can be individually cloned and expressed in the toxic yeast strain as previously described1,2,3,4.

Figure 1: PCR amplification of target gene. CT229 from Chlamydia trachomatis serovar L2 was PCR amplified from genomic DNA. PCR products were analyzed on a 1% agarose gel stained with ethidium bromide. Please click here to view a larger version of this figure.
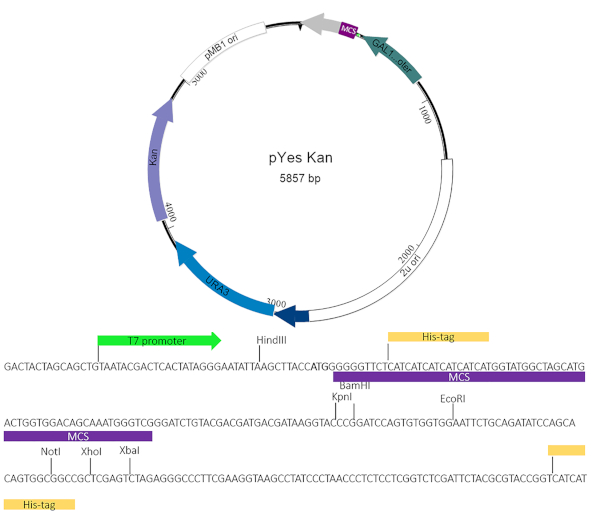
Figure 2: pYesNTA-Kan plasmid map. The target gene of interest was cloned into the multiple cloning site (MCS) of pYesNTA-Kan. Kanamycin was used for selection in E. coli and uracil dropout was used for selection in S. cerevisiae. Expression of the His-tagged fusion protein was verified by Western blotting. Please click here to view a larger version of this figure.
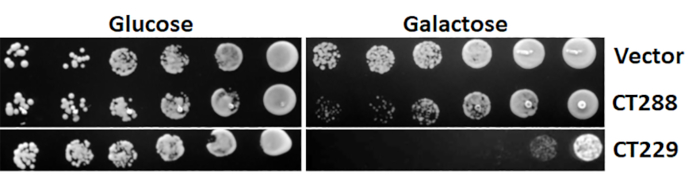
Figure 3: Representative yeast toxicity results. Effector proteins of interest were cloned into pYesNTA-Kan. Sequence-verified plasmids were transformed into S. cerevisiae and transformants were serially diluted and spotted on Ura– glucose and galactose agar to assess toxicity. Please click here to view a larger version of this figure.
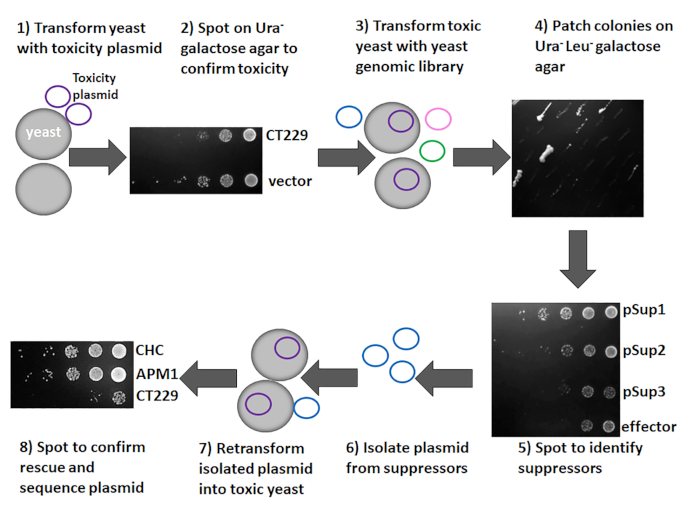
Figure 4: Overview of the yeast suppressor screen. The target gene of interest was cloned into pYesNTA-Kan and plasmids were transformed into S. cerevisiae. Transformants were serially diluted and spotted on Ura– glucose and galactose agar to assess toxicity. To identify the pathway targeted by the toxic effector protein, the yeast strain expressing the toxic protein was transformed with a yeast genomic library. Colonies were patched on Ura– Leu– galactose agar and spotted to assess toxicity. Plasmids were isolated from those that suppress the toxicity of the effector protein. Plasmids were retransformed into the toxic yeast strain to confirm that the isolated plasmid is a suppressor. Plasmids from suppressors were sequenced to identify the yeast ORFs present. Please click here to view a larger version of this figure.
Discussion
This protocol outlines step-by-step procedures for identifying host biological pathways targeted by bacterial effector proteins using a modified yeast toxicity and suppressor screen. The yeast strain used, S. cerevisiae W303, is auxotrophic for both uracil and leucine. Uracil auxotrophy of the strain is used to select yeast carrying the protein of interest on the pYesNTA-Kan vector while leucine auxotrophy is used to select for the yeast genomic library vector pYep13. The yeast genomic library plasmids carry 3−13 ORFs, so each ORF should be individually cloned into the p415-ADH vector6 or a similar vector and retransformed into the toxic yeast to identify which is the true suppressor. It is typical to identify several suppressors representing a host biological pathway. Suppressors can sometimes be direct binding partners of the bacterial protein of interest, but not always.
Generating yeast clones carrying the bacterial protein of interest on pYesNTA-Kan is the first major step and great care should be taken to preserve the integrity of these clones as soon as possible because there is strong negative selection for yeast carrying toxic proteins, even without intentional galactose induction, and they may lose the plasmid. Occasionally there is plasmid loss even when the yeast is maintained under selection on dropout media. A method has been previously reported to decrease the occurrence of plasmid loss by linearizing the vector and integrating it within the yeast genome1.
A major drawback of these techniques is that the suppressor screen only yields meaningful data if overexpression of the bacterial effector protein triggers an observable and consistent growth defect in yeast. The primary difficulty in this method is when no suppressors are identified. It is hard to determine whether a failure to identify suppressors is due to biological reasons or technical issues. From a biological perspective, there could be several causes: the protein of interest could be so toxic that proteins from the genomic library are not able to significantly suppress toxicity, the targeted pathway may be incapable of rescue, or numerous co-expressed factors may be required to overcome the toxic effect. Approaching the biological explanation is challenging, as there are many undefined variables to dissect. Because there is an established protocol, exploring technical explanations is a much more tractable approach. From a technical standpoint, a failure to identify suppressors is likely the result of low transformation efficiency of the yeast genomic library. This protocol uses the ATCC S. cerevisiae AB320 partially digested genomic library in pYEp13. Other libraries may be sufficient as well as other vectors, but this protocol specifically uses the ATCC library and there are no studies currently published that report alternate library or vector sources. It is strongly recommended that coverage of the yeast genome should be at least 10x, or underrepresentation may hinder the identification of suppressors. Decreased transformation efficiency can bring these numbers under 1x. Thus, parts of the yeast genome may not be represented in the suppressor screen. Low transformation efficiency can be due to many issues, including bad reagents or poor library preparation. This method largely adheres to the DMSO/LiAc transformation method first put forth by Hill et al.7. It is recommended that new DMSO, 50% PEG, and fresh herring sperm DNA are purchased and prepared for the transformations and used exclusively for these assays to decrease contamination or degradation of the quality of the reagents. DMSO is highly hygroscopic and absorbs water from the atmosphere readily. Therefore, making many individual use aliquots is recommended to avoid repeated opening of a container's lid and exposure to the atmosphere. High quality herring sperm DNA can be stored at -20 °C and remains fit for use for several years, although a fresh preparation should be prepared from the stock for each transformation. Once boiled and cooled, the herring sperm DNA should not be reboiled or reused, so it is best to only remove what is needed and avoid making more than is necessary for the current transformation. Attention to detail can greatly improve the transformation efficiency of yeast and improve the chances of identifying a suppressor.
Considering that the yeast pYep13 vector carries 3−13 different yeast ORFs, reducing the number of false-positive plasmids is paramount to identifying suppressors. Yeast can take up and harbor multiple copies of similar vectors, whereas the general wisdom associated with plasmid incompatibility traditionally dictates that E. coli only maintains one type of plasmid with the same origin of replication (ORI) at a time, although this is not always the case. This can become a major issue when attempting to identify suppressor candidates. If suppressor yeast clones are identified, the plasmids will be extracted and retransformed into E. coli for propagation and subsequent sequencing for identification. Once the candidate suppressor plasmid has been isolated and retransformed into E. coli, it is strongly recommended that at least five E. coli colonies are picked for sequencing, because it is possible that individual clones may be carrying different plasmids. Since E. coli should only be able to propagate one plasmid at a time, if the yeast isolation yields multiple plasmids, different E. coli clones on the plate should carry only one of them at a time. Therefore picking five colonies based on normal transformation efficiency in E. coli should show whether the clones are carrying the same or different plasmids. If multiple plasmids arise subsequent to sequencing of the plasmids from E. coli, then each one should be transformed into the toxic yeast clone and assessed for rescue of the growth defect. Furthermore, even if only a single suppressor plasmid is identified, for rigor and reproducibility retransformation of the plasmid into the toxic clone is encouraged.
The yeast suppressor assay is a very large experiment and appropriate time and materials should be planned accordingly. This protocol contains many details that must be adhered to strictly and it is recommended that the researcher conducting these assays carefully read and watch the protocol in full prior to starting an assay. This is a multipartite procedure including both bacteria and yeast that need to be cultivated at different temperatures. Isolation of plasmids from yeast can sometimes be difficult due to their tough cell wall and the use of DMSO helps achieve higher transformation efficiency.
One of the largest advantages of the yeast toxicity and suppressor screen is that the bacterial effector does not need to physically bind to the host substrates for any appreciable amount of time to detect the putative pathway targeted. Only a small handful of studies have reported using this or a similar technique1–5. However, there are reports of techniques that identify host pathways and many techniques to screen for binding partners of bacterial effector proteins. Kramer et al. reported a novel systems biology approach to identify pathways targeted by bacterial effector proteins8. The technique used a haploid deletion strain collection of S. cerevisiae to screen for mutants sensitive to the Shigella effector Osp4. Using this technique, Osp4 was linked to regulation of MAPK signaling and attenuation of innate immunity in host cells. Array-based, high-throughput, automated yeast-two hybrid screens can now rapidly identify putative bacterial effector binding partners by screening many at the same time against millions of host proteins9. Similarly, high-throughput immunoprecipitations coupled with mass spectrometry are being used to mass identify putative binding partners of entire families of bacterial effector proteins10. While many of these studies can accelerate the timetable of effector protein characterization, they rarely provide insight as to what physiological processes are being manipulated and what the holistic outcome of these interactions are. Thus, techniques such as the yeast toxicity and suppressor screen are essential for illuminating the cellular processes being manipulated by these effector proteins. At some point, observations identifying binding partners or pathways targeted by bacterial effector proteins need to return to cellular infection models to determine whether these in vitro findings hold true in physiologically relevant scenarios. Genetic manipulation of obligate intracellular pathogens like C. trachomatis has been a major hurdle until recently11–15 and generation of site-specific mutants to study effector proteins was not possible. In the last few years though, a revolution in generating chlamydial mutants, complementing them, and overexpressing target proteins has occurred. These recent advances have greatly enhanced the value of the information derived from studies using toxicity and suppressor screens as it is now possible to go from yeast directly into infection to probe the validity of the results.
Disclosures
The authors have nothing to disclose.
Acknowledgements
We thank Shelby Andersen, Abby McCullough, and Laurel Woods for their assistance with these techniques. This study was funded by startup funds from the University of Iowa Department of Microbiology and Immunology to Mary M. Weber.
Materials
| Agar | Fisher Scientific | BP2641500 | |
| Galactose | MilliporeSigma | G0750-1KG | |
| GeneJet Gel extraction kit | ThermoFisher Scientific | K0691 | |
| GeneJet PCR purification kit | ThermoFisher Scientific | K0701 | |
| GeneJet plasmid miniprep kit | Thermo | K0503 | |
| Glucose | MilliporeSigma | G8270-1KG | |
| Herring Sperm DNA | Promega | D1811 | |
| KpnI-HF | New England Biolabs | R3142S | |
| Lithium acetate dihydrate | MilliporeSigma | L6883-250G | |
| Peptone | Fisher Scientific | ||
| Phusion High-Fidelity DNA Polymerase | New England Biolabs | M0530 | |
| Poly(ethylene glycol) 3350 | MilliporeSigma | 1546547-1G | |
| pYep13 | ATCC | 37323 | |
| T4 DNA ligase | New England Biolabs | M0202S | |
| Tryptophan | MilliporeSigma | 470031-1G | |
| XhoI-HF | New England Biolabs | R0146S | |
| Yeast extract | Fisher Scientific | BP1422-500 | |
| Yeast miniprep kit | Zymo | D2001 | |
| Yeast nitrogen base without amino acids | MilliporeSigma | Y0626-250G | |
| Yeast Synthetic Drop-out Medium Supplements | MilliporeSigma | Y1501-20G | without uracil |
| Yeast Synthetic Drop-out Medium Supplements | MilliporeSigma | Y1771-20G | without uracil, leucine, tryptophan |
References
- Tan, Y., Luo, Z. Q. Legionella pneumophila SidD is a deAMPylase that modifies Rab1. Nature. 475 (7357), 506-509 (2011).
- Guo, Z., Stephenson, R., Qiu, J., Zheng, S., Luo, Z. A Legionella effector modulates host cytoskeletal structure by inhibiting actin polymerization. Microbes and Infection. 16 (3), 225-236 (2014).
- Tan, Y., Arnold, R. J., Luo, Z. -. Q. Legionella pneumophila regulates the small GTPase Rab1 activity by reversible phosphorylcholination. Proceedings of the National Academy of Sciences of the United States of America. 108 (52), 21212-21217 (2011).
- Weber, M. M., et al. The type IV secreted effector protein CirA stimulates the GTPase activity of RhoA and is required for virulence in a mouse model of Coxiella burnetii infection. Infection and Immunity. 84, (2016).
- Faris, R., et al. Chlamydia trachomatis CT229 subverts Rab GTPase-dependent CCV trafficking pathways to promote chlamydial infection. Cell Reports. 26, 3380-3390 (2019).
- Mumberg, D., Müller, R., Funk, M. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene. 156, 119 (1995).
- Hill, J., Donald, K. A. G., Griffiths, D. E. DMSO-enhanced whole cell yeast transformation. Nucleic Acids Research. 19 (20), 41070 (1991).
- Kramer, R. W., et al. Yeast Functional Genomic Screens Lead to Identification of a Role for a Bacterial Effector in Innate Immunity Regulation. PLoS Pathogens. 3 (2), 21 (2007).
- Häuser, R. T. S., Rajagopala, S. V., Uetz, P. Array-Based Yeast Two-Hybrid Screens: A Practical Guide. Two Hybrid Technologies: Methods and Protocols, Methods in Molecular Biology. , 21-38 (2012).
- Mirrashidi, K. M., et al. Global mapping of the inc-human interactome reveals that retromer restricts chlamydia infection. Cell Host and Microbe. 18 (1), 109-121 (2015).
- Wang, Y., et al. Development of a transformation system for chlamydia trachomatis: Restoration of glycogen biosynthesis by acquisition of a plasmid shuttle vector. PLoS Pathogens. 7 (9), 1002258 (2011).
- Mueller, K. E., Wolf, K., Fields, K. A. Gene deletion by fluorescence-reported allelic exchange mutagenesis in Chlamydia trachomatis. mBio. 7 (1), 1-9 (2016).
- Johnson, C. M., Specific Fisher, D. J. Site-Specific, Insertional Inactivation of incA in Chlamydia trachomatis Using a Group II Intron. PLoS ONE. 8 (12), 83989 (2013).
- Weber, M. M., et al. Absence of specific Chlamydia trachomatis inclusion membrane proteins triggers premature inclusion membrane lysis and host cell death. Cell Reports. 19 (7), 1406-1417 (2017).
- Weber, M. M., et al. A functional core of IncA is required for Chlamydia trachomatis inclusion fusion. Journal of Bacteriology. 198 (8), 1347-1355 (2016).
