Here, we present illustrative results from experiments where D. melanogaster was orally infected with P. aeruginosa or P. entomophila. Figure 2 demonstrates the successful oral infection of flies following a 12 h or 24 h exposure period to bacterial cultures of OD600 = 25 and 100 for P. aeruginosa (Figure 2A) and P. entomophila (Figure 2B, C), respectively. Figure 2B illustrates the importance of using a more concentrated culture of P. entomophila, shown by the increase in bacterial load when flies are exposed to bacterial cultures of greater optical density. Male and female Oregon R (OreR) flies clear P. aeruginosa infection at the same rate (Figure 3) and shed the same number of P. aeruginosa CFUs (Figure 4A). When infected with P. entomophila however, male and female OreR flies differ in the number of bacteria shed, in a manner that changes over time (Figure 4B). Males and females die from P. aeruginosa (Figure 5A) and P. entomophila (Figure 5B) at different rates. We also see that Dcy mutants (which lack the protective peritrophic matrix in the gut epithelium) and Relish mutants (which lack a functional IMD immune pathway), show decreased survival following P. entomophila and P. aeruginosa oral infection (Figure 5C).
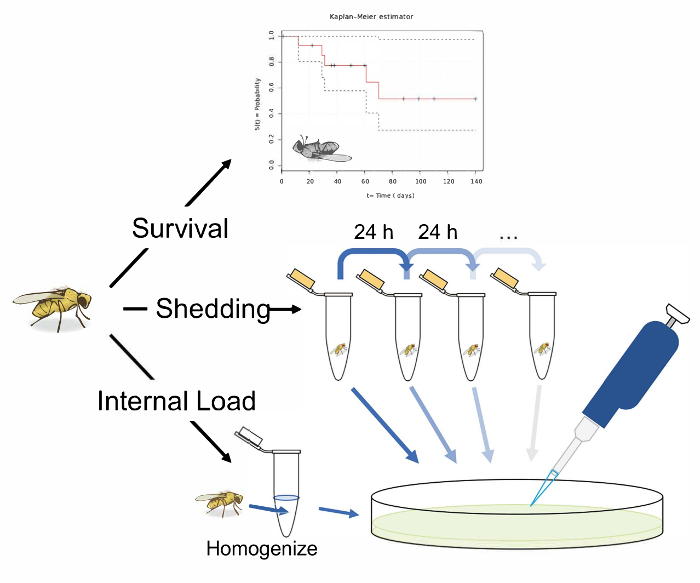
Figure 1: Schematic overview of protocols for measuring survival, shedding, and internal bacterial load following oral infection in Drosophila melanogaster. An illustration of 3 potential experiments following the oral infection of D. melanogaster. Measure the 'survival' by transferring single flies to vials and recording their infected lifespan. Measure 'shedding' by transferring single flies to 1.5 mL microcentrifuge tubes with 50 µL of Lewis medium in the cap. After 24 h in the tube, remove the fly and vortex the tube with 100 µL of 1x PBS. Remove and plate this solution on LB nutrient agar to calculate the bacterial shedding. Measure the shedding in the same fly longitudinally, by transferring flies to fresh tubes with Lewis medium in the cap after 24 h, and washing and plating the now contaminated tube. A fly's 'internal load' can be measured by taking an infected fly, surface sterilizing it, and homogenizing it before finally plating the homogenate on LB nutrient agar. This can be performed after shedding has been measured to calculate how the 'internal load' and shedding correlate. The fly illustration used in this figure was originally drawn by B. Nuhanen36. The authors have modified it to accompany the example Kaplan-Meier curve which is taken from Wikimedia Commons37. All other illustrations are original. Please click here to view a larger version of this figure.
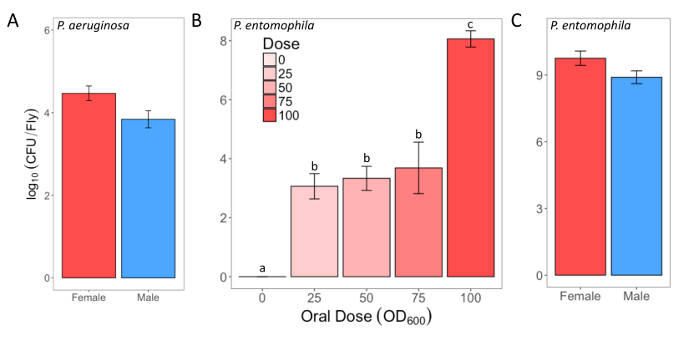
Figure 2: Infectious dose of bacteria following oral infection. (A) Infectious dose of male and female Oregon-R flies following exposure to a P. aeruginosa culture (OD600 = 25) for 12 h. The mean and SE were calculated from 3 males and 3 females. (B) Infectious dose of outcrossed wild-type females following exposure to one of four P. entomophila cultures (OD600 = 100, 75, 50, and 25) or control 5% sucrose solution for 24 h. The statistical difference of (F3,76 = 18.567, p <0.001) in the infectious dose between exposure treatments is denoted by differing letters above bars. The means were calculated from 5 flies for the OD600 = 0 dose, and 18-20 for all other doses. (C) The infectious dose of male and female Oregon-R flies following exposure to P. entomophila culture (OD600 = 100) for 24 h. The mean and SE were calculated from 20 males and 20 females. Please click here to view a larger version of this figure.
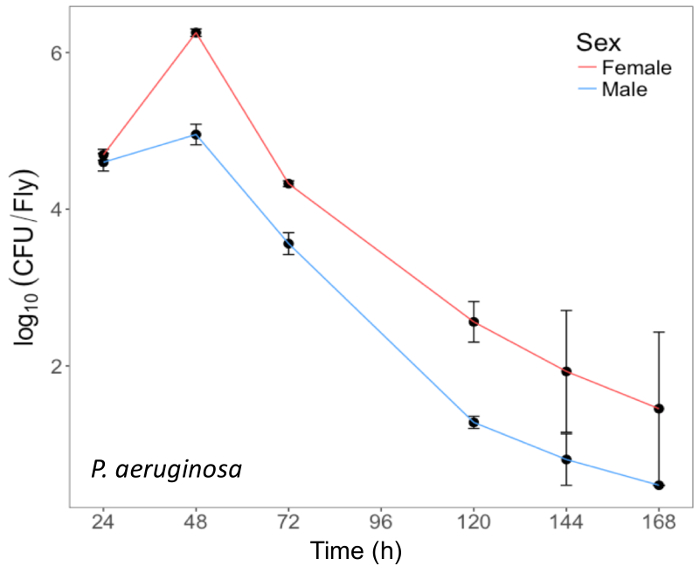
Figure 3: Internal P. aeruginosa load in flies after oral infection. Mean ± SE bacterial load of male and female Oregon-R flies following oral infection with P. aeruginosa (OD600 = 25) up to 168 h post-infection. The mean and SE of each time point are calculated from 3 individuals. A fly's internal bacterial load significantly changes over time (p <0.001). Please click here to view a larger version of this figure.
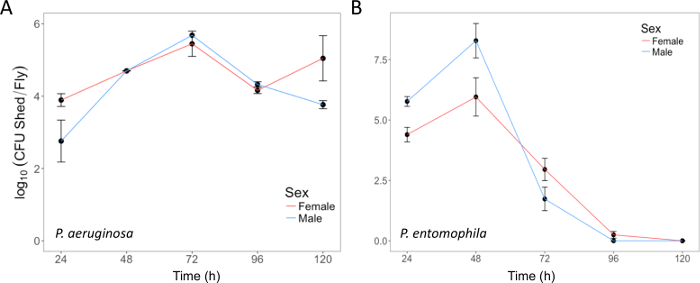
Figure 4: Bacterial shedding following oral infection. (A) P. aeruginosa shed by the same flies used in Figure 3, up to 120 h post-infection. The mean and SE were calculated from 3 males and 3 females. (B) P. entomophila shed by male and female Oregon-R flies following oral infection with P. entomophila (OD600 = 100) up to 120 h post-infection. The mean and SE were calculated from 34 males and 38 females. For both P. aeruginosa and P. entomophila, the number of CFUs shed by a fly significantly changes over time (p <0.001). Please click here to view a larger version of this figure.
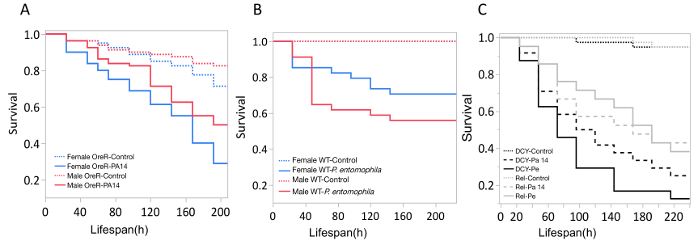
Figure 5: Survival of flies following bacterial oral infection. Kaplan-Meier (KM) survival curves of (A) Oregon-R male and female flies following oral infection with P. aeruginosa (OD600 = 25) or control 5% sucrose solution. The KM survival curve was calculated from 4 vials of 20 flies per treatment group. (B) OreR male and female flies after oral infection with P. entomophila (OD600 = 100). The KM survival curve was calculated from 4 single control flies and 34 infected flies for both males and females. (C) Immune mutants: Dcy (Drosocrystallin-peritrophic matrix mutant) and Rel (Relish-IMD mutant), exposed to P. entomophila (Pe), P. aeruginosa (Pa14), or a control 5% sucrose solution. All infected groups die significantly faster than the control flies (p <0.001). Please click here to view a larger version of this figure.
