Cell fractionation can be used to isolate and enrich membrane-associated proteins from cytosolic and nuclear proteins. Figure 1 is a Western blot demonstrating the contents of the three primary fractions that can be collected during the subcellular fractionation process. Specifically, Figure 1 shows that fractionation cleanly separates membrane proteins (i.e. Na+/K+ ATPase, protein disulfide isomerase (PDI), and HA-MOR1) from histone H2B and glyceraldehyde 3-phosphate dehydrogenase (GAPDH), nuclear proteins, and cytosolic proteins, respectively. Additionally, Figure 1 demonstrates the enrichment of proteins (lane 1 compared to lane 4) in their respective subcellular fractions as a result of fractionation. A representative Ponceau stain demonstrates equal protein loading in each fraction. It is important to note that this fractionation protocol does not distinguish between different cellular membranes. PDI is generally localized to the endoplasmic reticulum (ER), whereas Na+/K+ATPase is predominantly at the plasma membrane. However, both proteins are present in the final crude membrane fraction (Figure 1, lane 4). Additionally, while this protocol robustly separates nuclear proteins from the cytosolic and final membrane fraction (Figure 1, lanes 2-4), the fraction enriched for nuclear proteins contains some membrane proteins (Figure 1, lane 2), possibly from the ER.
Multiple pharmacological parameters can be derived to characterize a GPCR-ligand interaction via GTP-binding experiments (Table 1). For example, the half-maximal response (EC50) and Hill coefficient (nH) of an agonist can be derived by monitoring GTP binding in response to varying doses of the agonist. Figure 3A demonstrates dose-responsive GTP binding to MOR1 after DAMGO treatment. When the data is fit to a four-parameter nonlinear regression, the fit describes a receptor-ligand interaction with an EC50 of 185 ± 23 nM and a Hill coefficient of 0.46 ± 0.06. The shallow GTP-binding curve observed after DAMGO treatment suggests negative cooperativity between DAMGO and MOR1. This method can also identify and describe the pharmacology of an antagonist. As Figure 3A and B illustrates, naloxone is an MOR1 antagonist. The agonist potency (Kb) of naloxone, 97 ±20 nM, was determined by varying the concentration of naloxone in competition with a fixed concentration of DAMGO (Figure 3A). Naloxone exhibited a Hill coefficient of 0.88 ±0.06, suggesting independent binding between naloxone and MOR1. If the action of a ligand is unknown, this assay can discriminate between an agonist, antagonist, and inverse agonist. If the ligand is an agonist, there would be an increase in GTP binding, as in Figure 3A, following DAMGO application. If the ligand is an inverse agonist, there would be diminished binding of GTP relative to basal binding. If the ligand is an antagonist, there would be no effect upon treatment with ligand alone. If applied concomitantly with an agonist, an antagonist would inhibit the ability of the agonist to stimulate GTP binding. Figure 3A illustrates the antagonist activity of naloxone against the agonist DAMGO.
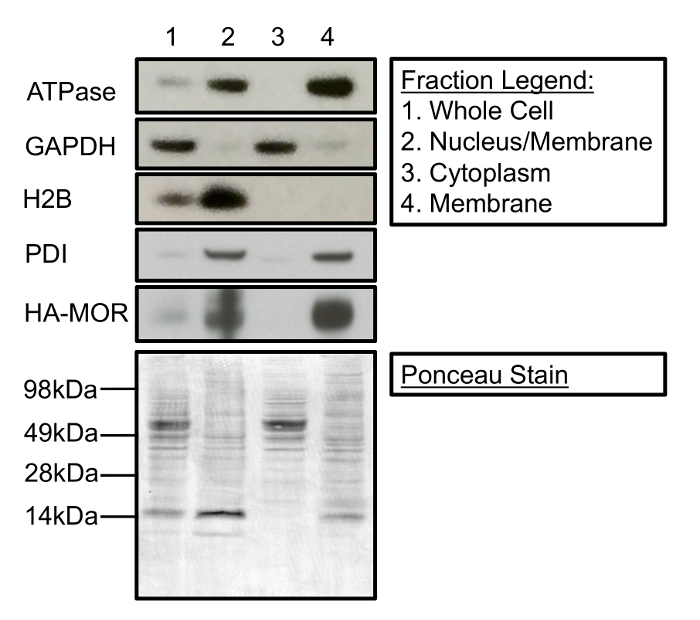
Figure 1: Cellular Fractionation Separates Membrane-associated, Nuclear, and Cytosolic proteins. (Top) Lane 1 represents the protein present in the whole cell. Lane 2 contains nuclear and membrane-associated proteins separated during the first centrifugation steps. Lane 3 is the cytosolic fraction separated following 20 min of centrifugation at 11,000 x g. Lane 4 contains a crude membrane fraction suitable for [35S]GTPγS-binding experiments. (Bottom) Ponceau stain of a Western membrane demonstrates protein loading for each cellular fraction. The following antibodies were used for immunoblotting: mouse anti-Na+/K+-ATPase (1:1,000), mouse anti-GAPDH (1:5,000), mouse anti-H2B (1:2,500), rabbit anti-PDI (1:1,000), and rat anti-HA (1:2,000). Please click here to view a larger version of this figure.

Figure 2: Flow Chart Outlining the Fractionation and [35S]GTPγS-binding Procedure. First, transfect the cells to express the GPCR of interest. After 48 h, harvest and fractionate the cells to isolate the receptor (red) and associated G-proteins (green = Gα, purple = Gβ, and orange = Gγ). To perform GTP-binding experiments, add [35S]GTPγS and incubate the membranes with the ligand of interest. To measure G-protein activity, bind the membranes to a filter to wash away any unbound radiochemical and then quantify the bound [35S]GTPγS by liquid scintillation counting. Please click here to view a larger version of this figure.
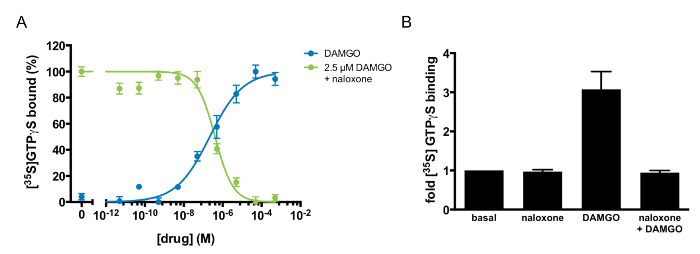
Figure 3: Agonism and Antagonism at µ-opioid Receptors Defined by [35S]GTPγS binding. (A) [35S]GTPγS dose-response curve to DAMGO alone (blue) or naloxone in competition with 2.5 µM DAMGO (green). [35S]GTPγS binding was normalized to the maximal stimulation in each experiment and was expressed as a percentage. The points shown are the mean of triplicate determinations and are expressed as the mean ±S.E.M. (B) Antagonism of DAMGO-stimulated [35S]GTPγS binding by naloxone. [35S]GTPγS binding was quantified after the addition of naloxone alone (100 µM), DAMGO alone (10 µM), or DAMGO (10 µM) in competition with naloxone (100 µM). The results are expressed as the mean ±S.E.M. of three independent experiments. Please click here to view a larger version of this figure.
| Ligand | EC50 or IC50 (nM) | nH | Kb (nM) |
| DAMGO | 185 ±23 | 0.46 ±0.06 | |
| Naloxone | 420 ±87 | 0.88 ±0.06 | 97 ±20 |
Table 1: Pharmacological Parameters of DAMGO and Naloxone Activity at µ-opioid Receptors. The half-maximal response (EC50) and Hill coefficient (nH) for DAMGO were derived from [35S]GTPγS binding in response to varying doses of DAMGO. The half-maximal inhibitory concentration (IC50) and equilibrium dissociation constant (Kb) for naloxone were determined from the effect of competition between naloxone and 2.5 µM DAMGO on [35S]GTPγS binding. The results are expressed as the mean ±S.E.M. of three independent experiments.
