We have used the protocol described above to measure the level of ER-mitochondria contacts upon the addition of three compounds known to inhibit specific GTPases. CDC42, RHO, and RAC are GTPases that promote actin polymerization28 when activated and are inhibited by ZCL278, Rhosin, and Ehop-016, respectively24. HEK293T cells transfected with split-Rluc were treated with DMSO (control), ZCL278 (50 µM), Rhosin (50 µM), or Ehop-016 (25 µM) and incubated for 1 h, 2 h, and 5 h. Using a plate reader, we measured split-Rluc activity at defined time points (Figure 2).
Rhosin (RHO GTPase inhibitor)-treated cells showed no significant changes in luciferase activity compared to the control DMSO-treated cells at 1 h, 2 h, and 5 h of the treatment (p > 0.9999, p= 0.6956, p > 0.9999) (Figure 2). These results demonstrate that Rhosin does not affect ER-mitochondria contacts, suggesting no involvement of RHO GTPase activity in regulating the contacts between the two organelles.
In contrast, the RAC GTPase inhibitor Ehop-016 showed significantly lower luciferase activity than DMSO at 1 h, 2 h, and 5 h of the treatment (p = 0.0106, p = 0.0009, p = 0.0024) (Figure 2). These data indicate that RAC GTPase activity is required for maintaining normal ER-mitochondria interaction in the cell.
Finally, CDC42 inhibitor ZCL278 showed the most drastic changes in luciferase activity (Figure 2). ZCL278-treated cells showed a significant decrease in luciferase activity compared to the control DMSO and had the lowest p-values through all three time points, maintaining a p < 0.0001 throughout. Our data demonstrate a strong inhibition of ER-mitochondria coupling when CDC42 GTPase activity is blocked by ZCL278, consistent with our previous report using the original split-Rluc assay performed in cells co-transfected with two vectors encoding each half of the split-luciferase separately24.
Given the striking results with ZCL278, especially when compared to the other GTPase inhibitors, ZCL278 activity was further tested at various concentrations [0.5 µM, 5 µM, 50 µM] against a DMSO control. Luciferase activities measured at 1 h, 2 h, and 5 h post treatment showed a dose-dependent response; 0.5 µM of ZCL278 resulted in the weakest effect on luciferase activity, while 5 µM showed stronger, and 50 µM showed the strongest reduction in reconstituted enzyme activity (Figure 3). Together, our results show that ZCL278 downregulates split-Rluc activity in a dose-dependent manner and, as time progresses, higher concentrations of ZCL278 (CDC42 inhibitor) still show a significant change relative to the control DMSO as compared to lower concentrations.
As a way to validate our split-Rluc assay, well-established independent assays, such as proximity ligation assay (PLA), transmission electron microscopy (TEM), and mitochondrial calcium uptake monitoring, can be performed (Figure 4 and data not shown)24. Isoproterenol, a β2 adrenergic receptor agonist, which is one of the compounds identified in our screen for ER-mitochondria contact promoters (Figure 4A), was used to confirm that the luciferase activity of the split-Rluc upon isoproterenol treatment was increased due to enhanced ER-mitochondria contacts as demonstrated by changes in PLA signal intensity as well as mitochondrial calcium uptake (Figure 4B-E).
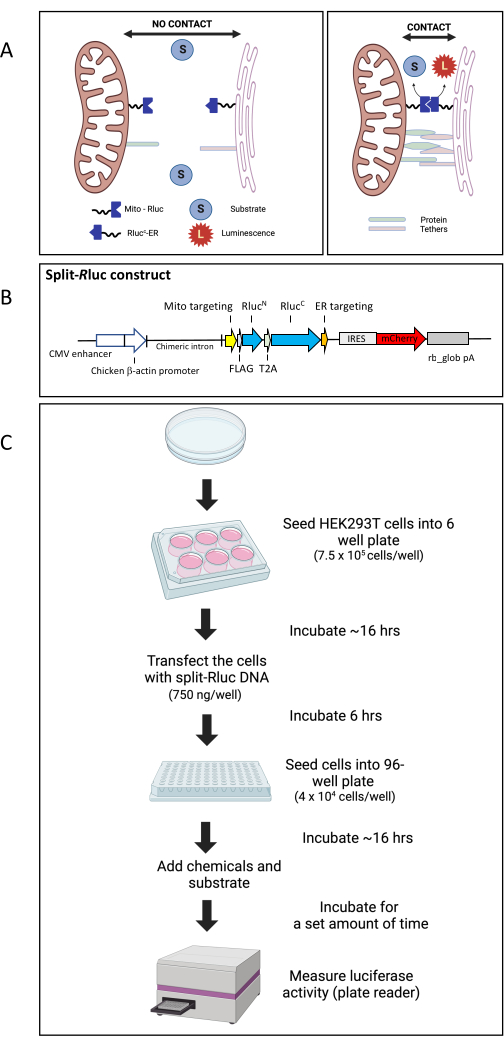
Figure 1: The split-Rluc reassembly assay. (A) Schematic representation of the Split-RLuc assay. Where there are no membrane contacts between the mitochondria and the endoplasmic reticulum, the Mito-RlucN and the RlucC-ER fail to interact and thus there is no Rluc8 activity. However, interaction between the Mito-RlucN and the RlucC-ER, where the ER and mitochondria membranes come into contact, results in full enzymatic activity, which is detected by the conversion of the substrate to luminescence. (B) Schematic map of the split-Rluc DNA construct (pCAG-MitoRlucN-T2A-RlucCER-IRES-mCherry). (C) Diagram of workflow for split-Rluc assay in HEK293T cells. Abbreviations: Mito = mitochondria; ER = endoplasmic reticulum; IRES, internal ribosomal entry site; rb_glob pA, rabbit beta-globin poly adenylation signal; T2A, a self-cleaving peptide 2A sequence from Thosea asigna virus. Please click here to view a larger version of this figure.
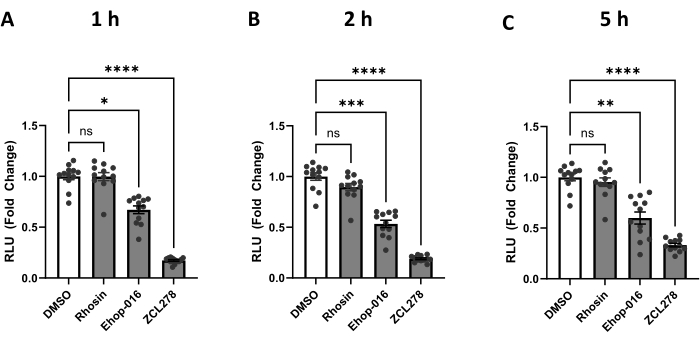
Figure 2: The split-Rluc activities corresponding to the levels of ER-Mito contacts in the cells treated with GTPase inhibitors. (A-C) Reconstituted luciferase activities at (A) 1 h, (B) 2 h, or (C) 5 h after DMSO (control), Rhosin [50 µM], Ehop-016 [25 µM], or ZCL278 [50 µM] treatment (n = 12 for each group). All data were analyzed using one-way ANOVA with multiple comparisons. P values are reported as p > 0.05 (ns), p ≤ 0.05 (*), p ≤ 0.01 (**), p ≤ 0.001 (***), p ≤ 0.0001 (****). Error bars indicate mean ± standard error of measurement. Abbreviations: Mito = mitochondria; ER = endoplasmic reticulum; RLU = relative light unit. Please click here to view a larger version of this figure.
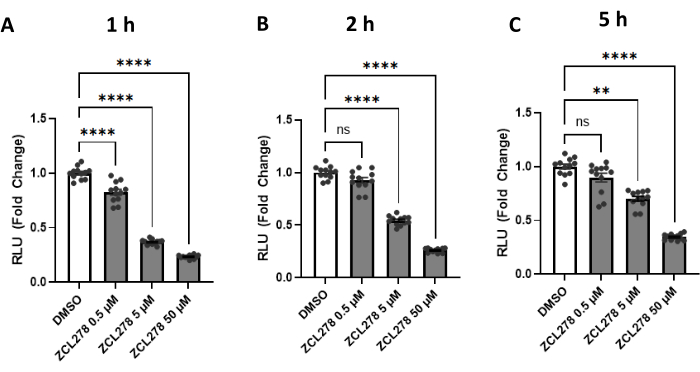
Figure 3: The split-Rluc activities measuring the levels of ER-Mito contacts in the cells treated with ZCL278. (A-C) Reconstituted luciferase activities at (A) 1 h, (B) 2 h, or (C) 5 h after DMSO (control) or ZCL278 at [0.5 µM], [5 µM], or [50 µM] concentration (n = 12 for each group). All data were analyzed using one-way ANOVA with multiple comparisons. P values are reported as p > 0.05 (ns), p ≤ 0.05 (*), p ≤ 0.01 (**), p ≤ 0.001 (***), p ≤ 0.0001 (****). Error bars indicate mean ± standard error of measurement. Abbreviations: Mito = mitochondria; ER = endoplasmic reticulum; RLU = relative light unit. Please click here to view a larger version of this figure.
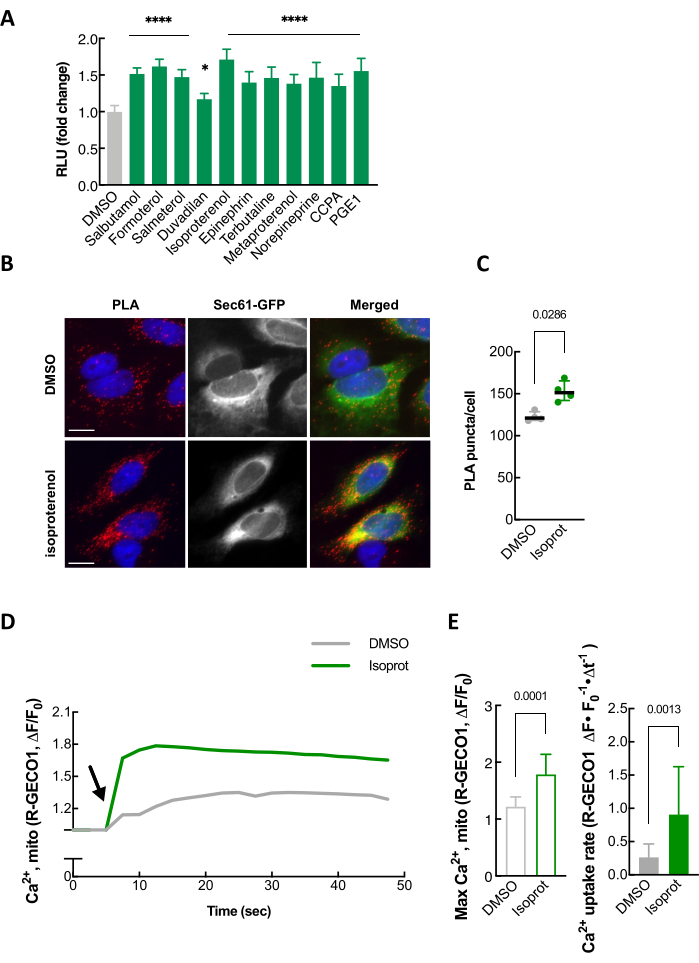
Figure 4: Validation of split-Rluc assay. (A) An example of quantification of split-Rluc activity after treating HEK293T cells with 11 drugs, including isoproterenol, identified in a compound screen for ER-mitochondria contact promoters. (B) An example of proximity ligation assay in HeLa cells that were treated with DMSO or isoproterenol (1 µM). PLA signal (red) indicates a close contact between ER and Mito. Sec61-GFP (green): to label ER; DAPI (blue): nucleus. (C) Quantification of PLA signal in A (n = 4 experiments with 77-97 cells each; Mann-Whitney test). (D) Mitochondrial Calcium uptake in response to histamine (100 µM). Mito-R-GECO1 fluorescent intensity change (average was taken over multiple HeLa cells) plotted every 2.5 s in DMSO- or isoproterenol-treated cells. Arrow indicates histamine addition. (E) Left: Maximum peak of mitochondrial calcium uptake in A. Unpaired two-tailed t-test (n = 10, DMOS; 14, Isoproterenol). Right: Mitochondrial calcium uptake rate (as in A; 7.5 to 12.5 s). Mann-Whitney test (n = 12, DMSO; 11, Isoproterenol). This figure is modified from Lim et al.24. Abbreviations: RLU = relative light unit; PLA = proximity ligation assay. Please click here to view a larger version of this figure.
Table 1: Summary of current techniques for ER-mitochondria contacts. List of advantages and limitations for each method. Please click here to download this Table.
Supplemental File 1: Zip file containing DNA sequence (PDF and GBK formats) of pCAG-MitoRlucN-T2A-RlucCER-IRES-mCherry. Please click here to download this File.
Supplemental Figure S1: Map of pCAG-MitoRlucN-T2A-RlucCER-IRES-mCherry. Please click here to download this File.
