All experiments were done in accordance with the Institutional Animal Care and Use Committees of the University of California, San Diego, and the Canadian Guide for the Care and Use of Laboratory Animals and were approved by the Université Laval Animal Protection Committee.
1. Alignment of the optical path between the CMOS (complementary metal oxide semiconductor) camera and the individual or branching patch cord
- Loosen all screws on the 5-axis translator (11, Figure 2B).
- Screw in the patch cord (12, Figure 2B) to the adaptor [SMA (sub-miniature A) or FC (fiber optic connector)] that is affixed to the 5-axis translator.
- Turn on the 470 nm excitation light (1, Figure 2B) at low power (100 µW), and place the tip of the patchcord pointing to an autofluorescent plastic slide. This does not have any bearing on future recordings but is solely for visualizing the alignment process.
- Record from the CMOS camera (13, Figure 2B) in live mode. Increase the gain or adjust the lookup table (LUT) until the image is not entirely black. The point is to be able to see an image at the focal point of the objective (10, Figure 2B).
- Advance the 5-axis translator towards the objective, ensuring that the 470 nm light is centered on the fiber at the SMA or FC end of the patch cord, until an image can be resolved on the camera.
- Adjust the X and Y axes until the image is centered and well-resolved.
- Visualize the light emitted from the ferrule-end of the patch cord. It should appear as an isotropic circle. If a branching patch cord is used, the amount of light emitted at the ferrule-ends of each patch cord should be similar. If the circle is not isotropic or the emitted light is unequal, adjust the 5-axis translator in the X-Y axis.
2. Setup of ROIs around fibers for measurement of mean fluorescent intensity
- Turn on all the excitation lights to better visualize the fibers. Adjust the camera gain such that no pixels are saturated and a clear image of the fibers are present.
- Live record or take a preliminary image.
- Draw ROIs around the fibers and keep them for the measurement of the mean intensity values during recordings (Figure 2A).
- For multiple fiber recordings, test for independence in signals.
- Live record from all fibers.
- Point one fiber towards a light source and tap with a finger. Very large fluctuations should occur solely in that channel (acceptable leakage 1:1000).
- If the signals are not independent, redraw more conservative ROIs and repeat the independence test.
- To label and keep track of which ROI corresponds to which fiber, colored tape or nail polish can be applied to the end of the fibers. Take a picture prior to the start of any experiment as a secondary reminder.
3. Setup of recording arena
- Hang the patch cord above the arena using stands, clamps, or holders.
- Make sure that the animal can freely move throughout the entire arena, uninhibited by the length of the fiber.
- Whether an operant box or open field is used, ensure that the patch cord will be able to reach the animal with minimal bending. If this requires a nose poke, ensure that there is enough room overhead to prevent bending of the fiber. Avoid any excessive bending or twisting of the patch cord.
4. In vivo recordings
NOTE: The procedure of optic fiber cannula implantation for fiber photometry experiments is identical to the procedure for optogenetics as described in Sparta et al15. We recommend using dental cement (see Table of Materials), which provides robust anchoring of the headcap to the skull bone. Dental cement will be particularly useful in cases where anchoring screws cannot be used.
- Visually inspect the distal end of the fibers of the patch cord by eye and with a minifiber microscope. If the surface of the fibers is scratched, repolish the fibers using fiber polishing/lapping film with fine grit (1 µm and 0.3 µm).
- Clean the distal ends of the patch cord with 70% ethanol and a cotton tip applicator.
- Clean the fiber-optic cannulas using 70% ethanol and a cotton tip applicator.
- Connect the ferrule end of the patch cord to the implanted fiber using a ceramic split-sleeve covered with a black shrink tube. During the connection, make sure that the sleeve is tight, otherwise use a new sleeve.
NOTE: There will be a large amount of signal loss if there is any space between the patch cord ferrule and the implant, and the recordings will not work. - Allow the animal to recover for a few minutes prior to the start of behavioral testing.
- Start recording the optical signal and run the experiment.
- While recording, keep a careful eye on the live-trace to ensure quality recordings. The signal is expected to rapidly decrease as a function of time in the first 2 min of recording. This effect is caused by heat-mediated LED decay, whereby the increase in heat increases the resistance of the optical element.
- If a jump in the signal that exceeds the on/off kinetics of GCaMP occurs, this is often an indication that the sleeve is not tight enough and the space between the patch cord and the implant is changing. In this case, stop the experiment and reconnect the animal using a new sleeve.
5. Fiber photometry data analysis
NOTE: This is a method for data analysis that works well for most recordings. However, alternative approaches can be implemented. Example code for data analysis can be found here: https://github.com/katemartian/Photometry_data_processing.
- Extract mean fluorescence intensity values recorded from 470 nm (Int470) and 410 nm (Int410) LEDs, corresponding to each individual fiber.
- Smooth each signal using a moving mean algorithm (Figure 3A).
- Perform baseline correction of each signal (Figure 3A and 3B) using the adaptive iteratively reweighted Penalized Least Squares (airPLS) algorithm (https://github.com/zmzhang/airPLS) to remove the slope and low frequency fluctuations in signals.
- Standardize each signal using the mean value and standard deviation (Figure 3C):
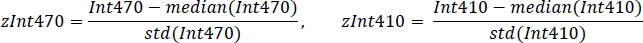
- Using non-negative robust linear regression, fit standardized zInt410 to zInt470 signals (Figure 3D) to the regression function:
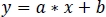
- Use the parameters of the linear regression (a, b) to find new values of zInt410 fitted to zInt470 (fitInt410, Figure 3D,E):
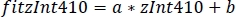
- Use the parameters of the linear regression (a, b) to find new values of zInt410 fitted to zInt470 (fitInt410, Figure 3D,E):
- Calculate the normalized dF/F (z dF/F) (Figure 3F):
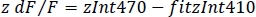
6. Simultaneous dual-color recordings
- Add to the photometry system a 560 nm LED to excite the red fluorescent calcium sensor and appropriate dichroic mirrors and filters (see Kim et al., 2016 for detailed description)12.
- Add an image splitter between the objective and the CMOS camera to separate the green and red emission wavelengths (see Figure 5). The image splitter will form two mirrored images on the camera sensor, corresponding to the red and green signals (e.g., a patch cord with 3 branches will create an image with 6 fibers).
- Draw ROIs around all fibers in both colors as detailed above. Make sure to clearly identify each ROI with the corresponding fiber and channel (green and red) (Figure 4A).
- Trigger simultaneous excitation with 470 nm and 560 nm LEDs and alternate them with 410 nm LED (Figure 5A).
7. Dual color data analysis
- Follow the steps in Section 5 to find fitInt410 for the Int470 signal and calculate z dF/F.
- Because the isosbestic point for red-shifted GECIs is generally unknown, the signal recorded with 410 nm LED in the green channel can be used for movement correction across both channels. Follow the steps in Section 5 to find fitInt410 for the Int560 signal and calculate z dF/F.
Neural correlates of behavioral responses can vary depending on a variety of factors. In this example, we used in vivo fiber photometry to measure the activity of axon terminals from the lateral hypothalamic area (LHA) that terminate in the lateral habenula (LHb). Wild type mice were injected with an adeno-associated virus (AAV) encoding GCaMP6s (AAV-hSyn-GCaMP6s) in the LHA and an optic fiber was implanted with the tip immediately above the LHb (Figure 4A). GCaMP6s expression is found in the cell bodies of the LHA and their axon terminals projecting to the LHb, where calcium signal can be recorded. Activation of the LHA-LHb pathway promotes passive avoidance in the real-time preference test suggesting that this pathway transmits aversive signals16. Mice were then connected to the fiber photometry system, placed in an open arena for 6 min, and exposed to 1 sec aversive airpuffs every 60 sec. The measured fluorescence significantly increased concurrently with the administration of airpuffs (Figure 4B-C). In mice expressing green fluorescent protein (GFP), no change in the signal was detected during the administration of airpuffs (Figure 4C). Following behavioral testing, the site of injection in the LHA and fiber placement above the LHb were confirmed histologically (Figure 4A).
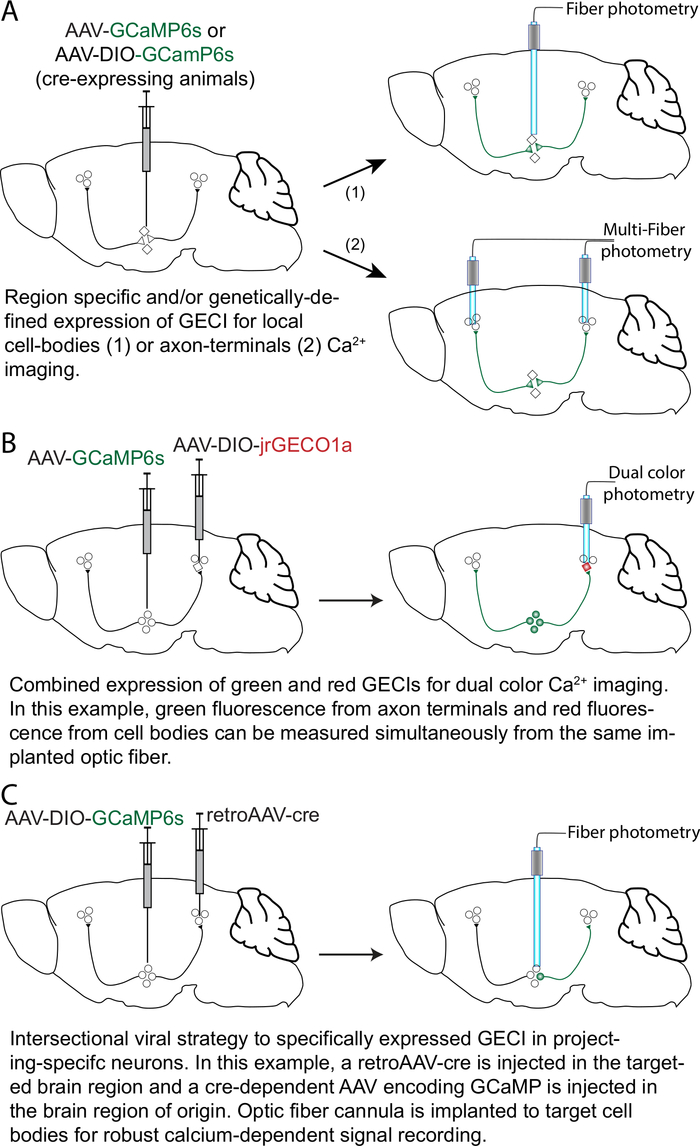
Figure 1: Strategies and approaches for GECI expression with anatomical and cell-type specificity. (A) A viral vector adeno-associated virus (AAV) encoding GCaMP6 (AAV-GCaMP6) is injected in a brain region of interest. The optic fiber can be chronically implanted with the tip placed over the cell bodies (1) or over the axon terminals (2). For selective expression in a genetically-defined neuronal population, a cre-dependent AAV (e.g., AAV-DIO-GCaMP6) can be injected in transgenic mice expressing the cre recombinase in a specific neuronal population. (B) For dual-color calcium imaging, in this example an AAV-GCaMP6s is injected in a brain region of interest, and a cre-dependent AAV encoding a red-shifted GECI (e.g., jrGECO1a; AAV-DIO-jrGECO1a) is injected in a genetically-defined neuronal population in a target brain region. The optic fiber is implanted for simultaneous calcium signal imaging of the axon terminals (green fluorescence) and cell bodies (red fluorescence). (C) For an intersectional viral strategy, a viral vector with retrograde transport properties (like retroAAV17) encoding the cre recombinase (retroAAV-cre) is injected in the target brain region together with an AAV-DIO-GCaMP6s injected in the projecting brain region in the same mouse. Optic fiber cannulas are implanted over the cell bodies for robust calcium-dependent signal recording. Please click here to view a larger version of this figure.
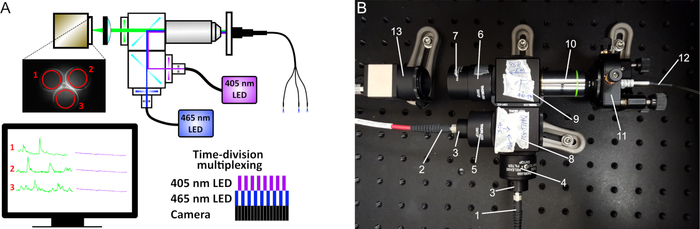
Figure 2: Fiber photometry schematic. (A) The excitation light from two LEDs (410 nm and 470 nm) passes through a series of filters and dichroic mirrors and produces an excitation spot at the working distance of the 20x objective. The light passes through either a single patch cord or bundled fibers (for multiple site recordings) that are connected to the implanted cannulas. The emitted fluorescence is collected by the same fibers, filtered, and projected on a CMOS camera sensor. On the captured images, the mean fluorescence intensity is recorded at the ROIs of each fiber. To simultaneously acquire signals from both 410 nm and 470 nm LEDs, a time-division multiplexing is implemented (lower right diagram). (B) Image of our custom-made photometry system and its components: (1) Fiber to the 465 nm LED, (2) Fiber to the 405 nm LED, (3) Collimators, (4) 470 nm bandpass filter, (5) 410 nm bandpass filter, (6) 535 nm bandpass filter, (7) Tube lens, (8) Cube with longpass 425 dichroic mirror, (9) Cube with longpass 495 dichroic mirror, (10) 20x objective, (11) 5-axis translator, (12) Mono- or bundled-fiber patch cord, (13) CMOS camera. Please click here to view a larger version of this figure.
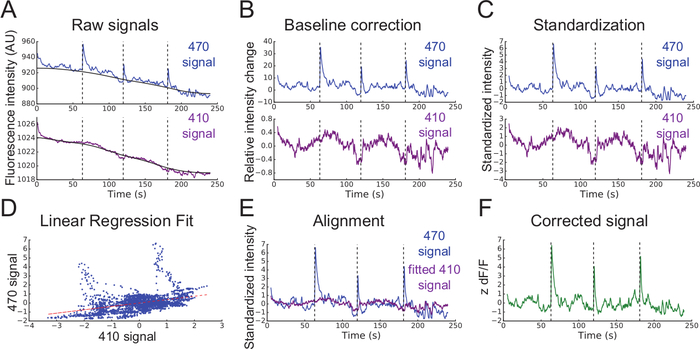
Figure 3: Analysis of fiber photometry data. (A) Smoothed mean fluorescent intensities (Int) recorded from 470 nm (top blue line) and 410 nm (bottom purple line) excitation wavelengths. Black lines are baselines found using the airPLS algorithm. (B) Relative intensity changes in signals after baseline correction. (C) Standardized 470 nm and 410 nm signals (zInt470, top; zInt410, bottom). (D) Non-negative robust linear fit of 470 nm and 410 nm signals. (E) Alignment of the trace Int410 to Int470 based on the fit. (F) Corrected and normalized calcium-dependent change in fluorescence (z dF/F). Please click here to view a larger version of this figure.
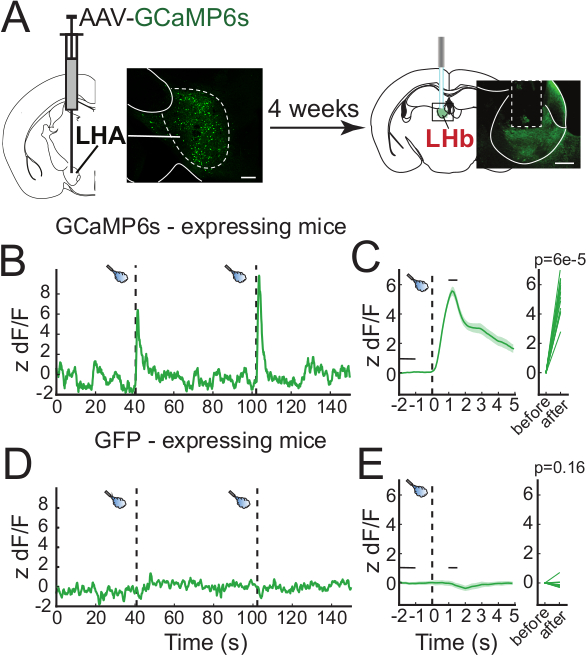
Figure 4: Representative results. (A) Diagram of the experimental procedure. An AAV-GCaMP6s is injected in the LHA of mice and 4 weeks later, an optic fiber cannula is implanted over the LHb for axon terminal signal recording. Inset are representative confocal images of GCaMP6s expression in the cell bodies of LHA neurons (left) and their axon terminals projecting to the LHb (right). (B) Representative calcium signal trace to airpuffs (dashed vertical bars) measured from LHb-projecting LHA axon terminals measured from a GCaMP6s-expressing mouse. (C) Peri-event plot of the average calcium response to airpuff events. The thick green line represents the average and the green-shaded regions represent the standard error of the mean (SEM, left panel), and the signal measured before and after an airpuff (3 mice, 15 events). (D,E) Same measurements as (B,C) for GFP-expressing mice (2 mice, 10 events). Scale bars are 200 µm. Please click here to view a larger version of this figure.
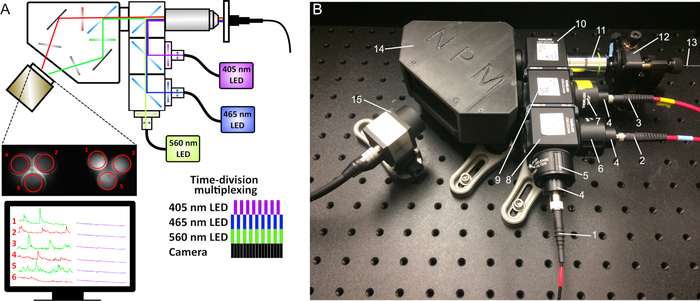
Figure 5: Schematic of dual-color fiber photometry. (A) An additional 560 nm LED, appropriate filters and dichroic mirrors, and an image splitter before the camera sensor were added to the original setup. (B) Photometry system components: (1) Fiber to the 560 nm LED, (2) Fiber to the 465 nm LED, (3) Fiber to the 405 nm LED, (4) Collimators, (5) 560 nm bandpass filter, (6) 470 nm bandpass filter, (7) 410 nm bandpass filter, (8) Cube with longpass 495 dichroic mirror, (9) Cube with longpass 425 dichroic mirror, (10) Cube with 493/574 dichroic mirror, (11) 20x objective, (12) 5-axis translator, (13) Mono- or bundled-fiber patch cord, (14) Image splitter, (15) CMOS camera. Please click here to view a larger version of this figure.
