The 13C nuclear magnetic resonance (NMR) spectrum of the prepared precursor molecule 3 displays the 12 sp-hybridized carbon atoms of the hexayne segment with the corresponding chemical shifts of δ = 82-60 ppm (Figure 1b). Moreover, the signals at δ = 173 ppm and at δ = 52 ppm are assigned to the carbonyl and methyl carbon of the ester, respectively. The signals between δ = 33-14 ppm are ascribed to the aliphatic carbons of the dodecyl residue. The corresponding UV/Vis absorption spectrum of 3 shows the characteristic vibronic fine structure of a hexayne (Figure 1c).
The film-forming properties of the hexayne amphiphile (3) are investigated by measurement of the surface pressure-area isotherm and by deriving the compressibility moduli (Figure 2a-b). Compression of the layer leads to an increase of the surface pressure at a mean molecular area of 24 Å2 and the isotherm features a steep slope. Moreover, a strongly tilted plateau region is observed between surface pressures of 9 and 15 mN/m, corresponding to mean molecular areas of 22 and 18 Å2, respectively. Above the plateau, a second steep increase of the slope of the isotherm is observed up to the collapse of the film at a surface pressure of 37 mN/m corresponding to a mean molecular area of 17 Å2. A plot of the compressibility modulus versus the surface pressure shows that the former increases to values of Cs-1 >100 mN/m even at surface pressures as low as 1-9 mN/m. This is followed by a decrease of the modulus in the plateau region, and a further increase beyond values of Cs-1 >300 mN/m up to the collapse of the film. Monitoring the layer of (3) at a surface pressure of 8 mN/m shows no change in the corresponding surface area (Figure 2c). At a surface pressure of 23 mN/m above the plateau in the isotherm, however, a significant reduction of the surface area occurs over the course of 45 min (Figure 2d).
The self-assembled monolayer at surface pressures below the plateau in the isotherm is characterized by recording infrared reflection absorption (IRRA) spectra (Figure 3). The IRRA spectra at surface pressures of 1-8 mN/m show broad bands at 3,600 and 1,670 cm-1 that arise from the OH stretching and bending vibrations of water, respectively. Moreover, a band at 2,350 cm-1 is observed that originates from an insufficient compensation of the carbon dioxide signal (Figure 3a). Next to these background signals, the spectra display bands at 2,919 and 2,849 cm-1 corresponding to the asymmetric and symmetric CH2 stretching vibrations of the dodecyl residues of amphiphile (3) (Figure 3b). The positions of these bands serve as qualitative markers for the conformational order of alkyl residues in monolayers at the air-water interface45,51. Moreover, bands at 2,200 and 2,171 cm-1 are observed that can be assigned to the C≡C stretching vibrations of the hexayne moiety (Figure 3c). The IRRA spectra do not change upon compression of the layer to different surface pressures below the plateau in the isotherm.
The carbonization process is monitored by IRRA spectroscopy and the evolution of the surface pressure is followed by means of the surface pressure microbalance (Figure 4). A comparison of the IRRA spectra before and after UV irradiation shows that the vibrational band associated with the hexayne moieties (ν(C≡C)) at 2,200 and 2,172 cm-1 has completely disappeared after 40 min of irradiation (Figure 4c). At the same time, the asymmetric and symmetric methylene stretching vibrations at 2,919 and 2,849 cm-1 decrease in intensity and shift to frequencies of 2,924 and 2,855 cm-1, respectively (Figure 4b). Moreover, the OH band of the subphase (ν(OH)) at 3,600 cm-1 significantly decreases in intensity in the course of irradiation (Figure 4a). The data from the surface pressure microbalance show that, when the barriers are fixed after compression of the monolayer to 8 mN/m and the UV irradiation is started, the surface pressure rapidly increases beyond values of 27 mN/m in the course of the carbonization process.
Films transferred to sapphire substrates after the UV-induced carbonization show a broad, featureless absorption at wavelengths of up to 550 nm in the respective UV/Vis spectrum (Figure 4e). The main absorption peak of the carbon nanosheets is observed at approximately 260 nm, and the comparison with the spectrum of the monolayer before irradiation confirms the complete conversion of the hexayne moieties. Brewster angle micrographs (Figure 5) and scanning electron microscopy (SEM) images serve as a means to visualize the obtained carbon nanosheets (Figure 6). While Brewster angle micrographs of a monolayer of the amphiphile (3) compressed to 8 mN/m show a film with imperfections or voids as indicated by the black regions (Figure 5a), the images of the layer obtained after carbonization by UV irradiation display a distinct change in the texture of the film (Figure 5b, c). After rupture of the sheet islands remain floating at the air-water interface (Figure 5d). Electron microscopy shows nanosheets after Langmuir-Schäfer transfer to a holey carbon TEM grid as support. The nanosheets are mechanically stable enough to span the micrometer-sized holes in a TEM grid (Figure 6a-d). A slight contrast is observed between regions covered with the carbon nanosheet and others at an accelerating voltage of 2.0 kV (Figure 6a; the dark spot visible in the image arises from the small working distance leading to a shadowing effect at low magnifications). Images taken at the same position highlight the influence of the accelerating voltage, as the carbon nanosheet becomes opaque to the electron beam at around 0.5 kV, and the carbon nanosheet shows draping as well as wrinkles at its edge (Figure 6b, c). The film is otherwise very smooth and uniformly spread over the grid further away from the border region (Figure 6d; the defect in the supporting grid aids in identifying the carbon nanosheet).

Figure 1: (a) Synthesis of the hexayne amphiphile (3) by the sequential bromination52,53 and Pd-catalyzed elongation30,31 of the alkyne segment. Reagents and conditions: (i) 1,4-bis(trimethylsilyl)buta-1,3-diyne, MeLi · LiBr, ZnCl2, PdCl2(dppf) · DCM, THF/toluene, 71%; ii) AgF, NBS, MeCN; then 1-trimethylsilyloctadeca-1,3,5-triyne 7, MeLi · LiBr, ZnCl2, PdCl2(dppf) · DCM, THF/toluene, 23% over two steps; (iii) NaOMe, DCM, MeOH, quantitative. (b) The 13C NMR spectrum of the hexayne amphiphile (3) with twelve acetylene carbon resonances (orange) and (c) the corresponding UV/Vis spectrum. Please click here to view a larger version of this figure.

Figure 2: Investigation of the hexayne amphiphile (3) at the air-water interface. (a) The surface pressure area isotherm and (b) the plot of the compressibility modulus of the film indicate a direct transition from a gas-analogous phase to a condensed phase. (c) A layer is compressed to 8 mN/m as well as (d) 23 mN/m, and the development of the surface area is monitored at constant surface pressure. Please click here to view a larger version of this figure.
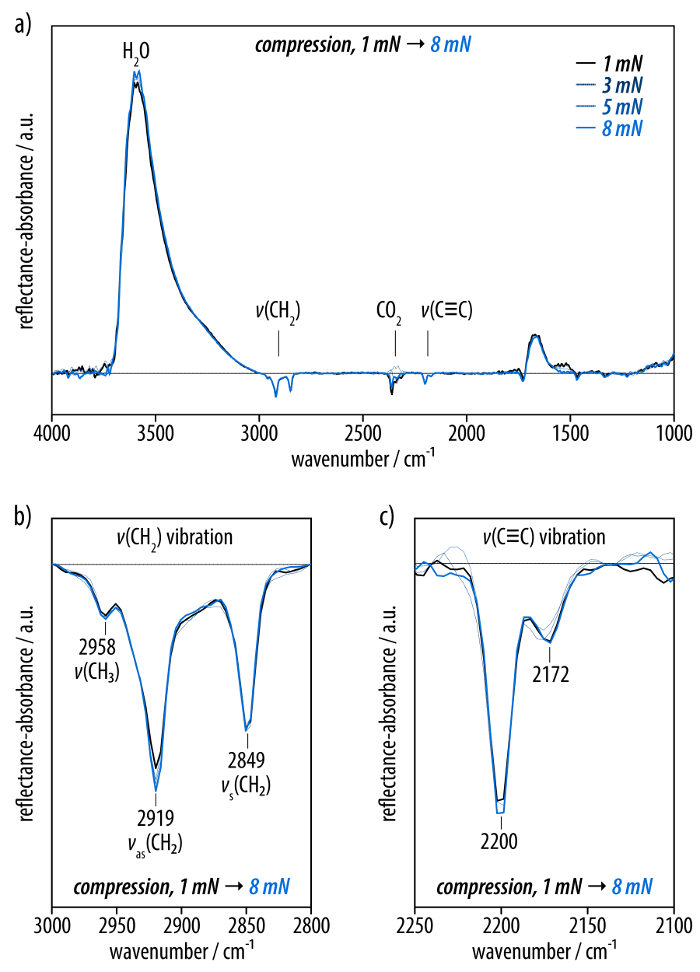
Figure 3: Infrared reflection-absorption (IRRA) spectra of the film of the hexayne amphiphile (3) (40°, p-polarized light) compressed to surface pressures between 1 mN/m (black line) and 8 mN/m (light blue line). (a) The full spectrum with prominent bands at 3,600 and 1,670 cm-1 from the water subphase as well as the peak around 2,350 cm-1 due to insufficient carbon dioxide compensation. (b) The spectral region of the methylene stretching vibrations as well as (c) the bands corresponding to the hexayne moiety. Please click here to view a larger version of this figure.
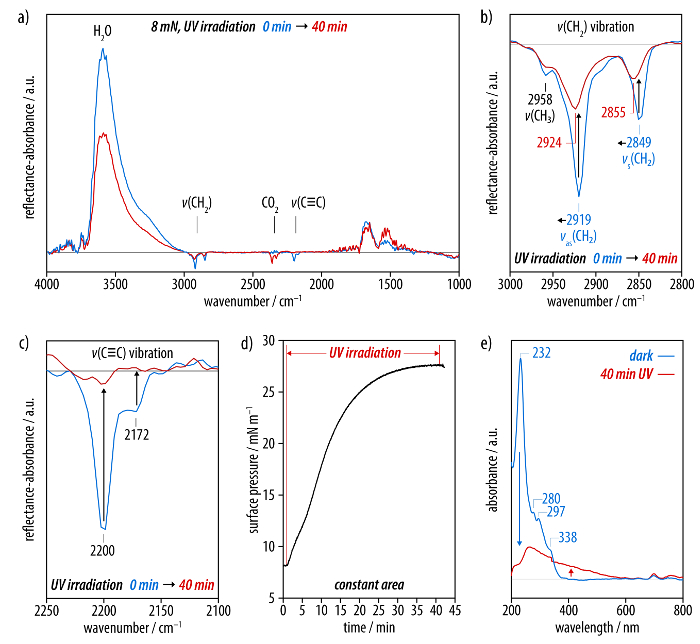
Figure 4: Investigation of the carbonization of a film of the reactive, carbon-rich amphiphile 3. (a) IRRA spectra recorded before (blue line) and after 40 min (red line) of UV irradiation. (b) The spectral region of the methylene stretching vibrations as well as (c) the bands corresponding to the hexayne moiety. (d) With the barriers fixed to a constant surface area, a significant increase in the surface pressure is observed during the carbonization. (e) UV/Vis spectra of irradiated films in comparison to a non-carbonized film of (3) show a broad and featureless absorption at wavelengths of up to 600 nm. Please click here to view a larger version of this figure.

Figure 5: Brewster angle microscopy (BAM) experiments with a film of amphiphile 3 at the air-water interface before and after carbonization by UV irradiation. (a) Micrograph of a monolayer of (3) compressed to 8 mN/m. (b) After UV irradiation, a clear change in the texture of the film is observed that (c) becomes more homogenous after allowing the film to expand by opening the barriers. (d) Rupture of the carbonized sheet by manipulation with a needle leaves islands floating at the air-water interface. Please click here to view a larger version of this figure.

Figure 6: Scanning electron microscopy (SEM) of a carbon nanosheet after Langmuir-Schäfer transfer to a holey carbon TEM grid as support. (a) A partially covered grid imaged at an accelerating voltage of 2.0 kV. (b–c) The carbon nanosheet becomes opaque to the electron beam at around 0.5 kV. (d) Away from the edge, a smooth film uniformly spreads the grid. Please click here to view a larger version of this figure.
