All appropriate guidelines and standard microbiological, safety, and cell culture practices were employed for the use and handling of the RG2 pathogen Leishmania major and recombinant DNA. All experiments with live L. major were performed in a biosafety cabinet in a BSL-2 certified laboratory. The work was overseen by the Texas Tech University Institutional Biosafety Committee.
NOTE: From a safety perspective, live L. major promastigotes are Risk Group 2 pathogens. Handle using appropriate containment, precautions, and oversight from the Institutional Biosafety Committee (IBC). Handle toxic substances and chemicals in accordance with institutional procedures for toxic substances. If recombinant toxins are used, IBC approval and oversight may be needed for recombinant DNA work.
1. Cultivation and preparation of L. major promastigotes
- Obtain, or make and validate, L. major genetic mutants as previously described using either homologous recombination or CRISPR-based methods13,21. Use knockouts complemented with the gene added back on a plasmid to ensure specificity of the knockout.
- Culture wild type L. major and spt2– promastigotes at 27 °C in complete M199 medium. Culture the episomal addback cells (spt2–/+SPT2) in complete M199 plus 10 µg/mL G418 (see Table 1 and Table of Materials).
NOTE: The entire experimental setup involving experiments with L. major cells must be performed in a BSL2-certified biosafety cabinet. - Cultivate the promastigotes in complete M199 medium22 until they reach log phase (2-8 x 106 cells/mL), as determined by L. major growth curve assays done previously23. Plan 1 x 105 cells for each well for cytotoxicity, plus 5 x 105 cells for staining control. For western blot, plan 2 x 107 cells per well.
NOTE: Perform cytotoxicity assay with two technical replicates. - To verify the proper cell density, mix an aliquot (10-40 µL) of promastigotes with an equal volume of fixative (3.7% paraformaldehyde in 1x PBS). Load 10 µL of the fixed sample onto each side of the hemacytometer.
CAUTION: Formaldehyde is a toxic chemical. Handle in accordance with institutional policies for hazardous chemicals. - Perform cell counting using a microscope at 20x magnification. Count all the cells in the 25 small squares in the center of the hemacytometer. Repeat for the squares on both sides and average the counts.
NOTE: If the variation between counts is >10, recount and average. If the average count is <10 or >100, alter the dilution and recount, and then calculate the culture density using the following formula:
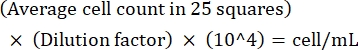 (Eq 1)
(Eq 1)
For example, if there are on an average 250 Leishmania in 25 squares, the culture density is 5 x 106 cells/mL. - After counting, transfer 5 x 106 cells to a 15 mL conical tube and centrifuge at 1,500 x g for 8 min at room temperature to pellet the cells.
- Discard the supernatant using a 10 mL pipette and briefly vortex the cell pellet. Add 5 mL of 1x PBS to the same tube and wash the cells by gently inverting 3-6x. Centrifuge at 1,500 x g for 8 min at room temperature to pellet the cells.
- Discard the supernatant using a 10 mL pipette and resuspend the 5 x 106 cell pellet in 5 mL of medium (e.g., M199 or 1x Tyrode's buffer) used for the experiments with a 5 mL pipette to give a final concentration of 1 x 106 cells/mL.
2. Cytotoxicity assay
- Experimental preparation
- Purify the toxin as previously described24, or purchase the toxin from a vendor. Aliquot into single-use aliquots and store −80 °C for up to 1 year. Avoid multiple freeze-thaw cycles.
- Determine the hemolytic activity for each toxin using human red blood cells (see Table of Materials)24.
NOTE: Hemolytic activity is used because it controls for differences in activity due to purification, mutations, etc. The species choice of erythrocytes may alter the hemolytic activity (e.g., intermedilysin requires human red blood cells). - Plan two technical replicates for each condition, seven dilutions for the dose-response curve, and a no-toxin control.
NOTE: With wild-type (WT), spt2–, and spt2–/+SPT2 promastigotes, two treatments may be tested in one V-bottom 96-well plate. For example, sensitivity to media could be compared (Figure 1). Instead of a V-bottom plate, 1.2 mL microtiter tubes (see Table of Materials) may be used. Due to the acquisition times on the cytometer, running more than one plate at a time is not recommended. - Determine which assay buffer to use based on the test conditions needed and the purpose of the experiment.
NOTE: In this example, two assay buffers are compared: M199 and Tyrode's buffer supplemented with the viability dye propidium iodide (PI). Cholesterol in serum will interfere with CDC activity24. - Calculate the amount of toxin needed based on the conditions and number of genotypes treated. Ensure that 50% specific lysis occurs halfway down the dilution curve.
NOTE: For CDCs, a two-fold serial dilution will give a good range for later logistic modeling. For spt2– promastigotes, 4,000 HU/mL SLO is the recommended starting dilution. Where inactive toxins are used, a mass equivalent to the highest dose may be used instead. - Plan a 200 µL final volume per well, and add a small volume extra (50-100 µL) to account for pipette error.
NOTE: With three genotypes, each done in duplicate, there will be six samples for the serial dilution. - Determine the total amount of toxin needed using the following formula:
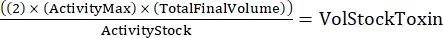 (Eq 2)
(Eq 2)
where ActivityMax is the highest concentration used (HU/mL); TotalFinalVolume is the total volume (for six samples, 200 x 6 + 100 = 1,300 µL); ActivityStock is the activity of the toxin stock (HU/mL); and VolStockToxin is the volume of toxin stock needed. - Prepare sufficient assay buffer needed for the experiment. Supplement the basal medium with a viability dye and any Ca2+ or EGTA needed to control Ca2+ levels. Vortex to mix.
NOTE: For example, for every 10 mL Tyrode's buffer, add 50 µL of 2 mg/mL PI and 200 µL of 100 mM CaCl2, giving final concentrations of 10 µg/mL and 2 mM, respectively. - Ensure the viability dye PI does not conflict with any other probes used, such as for fluorescent binding assays using Cy5 or AlexaFluor647-conjugated toxins20,25.
- Plan toxin dilutions to make a 2x solution of toxin. Add assay buffer to 1.5 mL centrifuge tubes and chill on ice. Only add the toxin immediately prior to starting the assay.
NOTE: In this example, 1.3 mL of assay buffer would be added for the top dilution, and 650 µL would be added for serial dilutions to prepare the 2x toxin solution.
- Experiment
- Reserve 0.5 mL of processed promastigotes from step 1.8 in a separate tube as "Unstained control".
NOTE: This sample will be used to set up gating on the flow cytometer. - Add 2 mg/mL PI to a final concentration of 10 µg/mL to the remaining promastigotes. Vortex for 3 s.
NOTE: After the addition of PI to the processed promastigotes, these cells can only be used for a time period of 2.5 h. After 2.5 h, the cells start to die, and the results become erroneous. - Add 1 x 105 (in 100 µL/well) processed promastigotes to each well of a V-bottom 96-well plate or 1.2 mL microtiter tubes (Figure 1). Place the plate or tube rack on ice at an approximately 45° angle from viewing. Perform the work in a biosafety cabinet when handling Leishmania promastigotes.
- Add 100 µL of assay buffer with PI to each no-toxin control (last row). Verify that the control was correctly added by visually identifying tubes with a total volume of 200 µL that appear darker in color.
- Remove the toxin aliquots from −80°C, thaw on ice, and pool as needed. Add the volume of toxin calculated in step 2.1.5 to the highest dilution (prepared in step 2.1.10). Then, serially dilute the toxin (Figure 1). Pipette up and down at least 8x to ensure mixing.
NOTE: Perform on ice because CDCs rapidly inactivate at room temperature. - Starting from the lowest toxin concentration, quickly add 100 µL of toxin to the correct row (Figure 1 and Figure 2) and continue until all the toxin has been added to the cells.
- Seal the plate with sealing tape. Incubate at 37 °C for 30 min. After the incubation period, package and transport the plate to the flow cytometer.
- Reserve 0.5 mL of processed promastigotes from step 1.8 in a separate tube as "Unstained control".
- Data acquisition
- Set up the flow cytometer (see Table of Materials) and acquisition software according to the manufacturer's instructions and per facility policy. Do not perform the flow cytometry procedure without prior training on the cytometer.
NOTE: In this example, a 4-laser Attune NxT was used. PI was collected on the YL-1 channel (excited by a 561 nm laser, passed through a 577 LP, reflected from 600 DLP, and filtered through 585/16 bandpass), though the wide spectrum of PI allows collection on other channels. - Using the unstained L. major promastigote sample, set the gates for forward and side scatter and the initial fluorescent parameters based on the dyes chosen.
- Include one extra parameter if desired for dot plots to check autofluorescence (e.g., "no stain") (Figure 3).
NOTE: In this example, the BL-1 channel (excited by a 488 nm laser, passed through 495 DLP and 503 LP, reflected from 555 DLP, and filtered via 530/30 bandpass) was used. - Using single stained controls, set the gates for the viability dye (PI in this study) and any fluorescently labeled toxins. Monitor forward scatter versus time for micro-clogs.
NOTE: PI will dimly stain all the cells above unstained controls. Dead cells will be readily separable, with transiently permeabilized cells in between populations. - Acquire >10,000 gated events for each sample on the cytometer.
NOTE: It is recommended to read from most sensitive to least sensitive, but the order of acquisition can be reversed to determine any impact of read order on sample results. - Save the data and export as needed for analysis.
- Set up the flow cytometer (see Table of Materials) and acquisition software according to the manufacturer's instructions and per facility policy. Do not perform the flow cytometry procedure without prior training on the cytometer.
- Data analysis
NOTE: In this study, Excel with Solver plug-in (see Table of Materials) was used for data analysis (see Supplemental File 1).- Gate total, single cell L. major promastigotes by gating on forward and side scatter and time as needed (Figure 3). Use height or area as recommended for the flow cytometer.
NOTE: For the flow cytometer used here, height is the recommended parameter instead of area. - Identify and gate dead cells as "PI high". Gate intermediate cells as "PI low". PI high cells are dead cells, while PI low cells are transiently permeabilized26.
NOTE: PI high cells typically show a 2-3 log shift from negative cells. - Export the data to Excel. Obtain the sample name/ID and %PI high to determine killing.
NOTE: If fluorescent toxins were used, the median fluorescent intensity (MFI) of live, transiently permeabilized, and negative populations will be needed. %PI low may be exported for transient permeabilization. - Determine the average %PI high for each condition between the two technical replicates.
NOTE: If average MFI is needed for fluorescent toxins, calculate this as well. - Calculate %Specific Lysis from the %PI high using the following formula24,25:
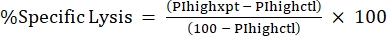 (Eq 3)
(Eq 3)
where PIhighxpt is the %PI high for the experimental condition; and PIhighctl is the %PI high for the no-toxin control. - Plot %Specific Lysis against toxin concentration for the dose-response curve (Figure 4).
- Organize the dose-response curve in Excel for logistic modeling. Include toxin concentration and average %Specific Lysis along with experimental details and/or raw %PI high calculations (Table 2).
- Verify that the Solver add-in is enabled.
NOTE: To enable the Solver in the desktop version of Excel, go to File > Options > Add-ins, and check the Solver box. Restart Excel. - Label four more columns as "modeled", "residuals", "parameters", and "parameter values". Verify that the first columns correspond to the experimental parameters, the toxin concentration, and %Specific Lysis (Table 2).
- Add the following parameters in the "parameters" column: L, k, c, SUM, and LC50. Initialize the parameters L, k, and c by entering the following values in the "parameter values" column: 100, 0.05, 1,000.
- In the "modeled" column, create the logistic model using the following formula:
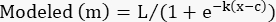 (Eq 4)
(Eq 4)
Set L, k, and c to the cells containing those parameters in the "parameter values" column.
Set x to the cell containing the toxin concentration.
NOTE: For Table 2, cell G4, the formula is as follows: =$J$3/(1+EXP(-$J$4*(D4-$J$5))) - Apply this equation to all the %Specific Lysis values except the no-toxin control.
- In the "residuals" column, calculate the square of the difference between the modeled number and the actual specific lysis using the following equation:
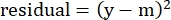 (Eq. 5)
(Eq. 5)
where y is the experimental %Specific Lysis; and m is the corresponding value in the "modeled" column calculated in steps 2.4.10-2.4.11. - In the "parameter values" column next to "SUM", sum all the values in the residuals column.
- In the "parameter values" column next to "LC50", initialize the equation to calculate the LC50 from the determined values. This is solving Eq 4 for x when m = 50.
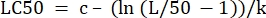 (Eq 6)
(Eq 6)
For Table 2, the Excel formula is as follows: =J5-(LN(J3/(50)-1))/J4 - Open the Solver from the Data Tab. Select the Set Objective to be the cell containing the sum of the residuals calculated. Set it to Min.
- Change the variable cells for the parameter values of L, k, and c.
NOTE: The solver may behave better if "Make Unconstrained Variables Non-negative" is left checked. - For negative k values, modify Eq 4 and Eq 6 by factoring out −1 from k to change k to positive. Use the GRG Nonlinear solving method. Click Solve.
- Check the curve and that the LC50 is automatically calculated using Eq 6 (Table 2). Verify the fit by graphically plotting both %Specific lysis and modeling against toxin concentration.
NOTE: It may also be checked by calculating the R2 for the curve.
- Gate total, single cell L. major promastigotes by gating on forward and side scatter and time as needed (Figure 3). Use height or area as recommended for the flow cytometer.
3. Protein analysis of toxin-challenged L. major promastigotes
- Prepare L. major promastigotes as described in section 1.
- Resuspend 2 x 107 WT, spt2–, and spt2–/+SPT2 L. major promastigotes in 2 mL of the desired assay buffer using a 5 mL pipette (e.g., serum-free M199). Add toxin to a final sublytic concentration and incubate at 37 °C for 30 min.
NOTE: For example, a sublytic dose of SLO for spt2– promastigotes is 500 HU/mL. - Include other genotypes and no-toxin controls.
- Centrifuge the promastigotes at 1,500 x g for 10 min to pellet the cells. Discard the supernatant using a 10 mL pipette. Run the closed tube containing the cell pellet rapidly three times across an irregular surface, such as the grill of the biosafety cabinet, to break up the cell pellet.
NOTE: The cell pellet is almost invisible to the naked eye. - Reconstitute 1x SDS-PAGE sample buffer with 2-mercaptoethanol immediately before use and heat to 95 °C for 10 min before addition to the cell pellet. Resuspend the cell pellet in hot 1x SDS-PAGE sample buffer, and mix well by pipetting up and down. Heat the resuspended cell pellet in 1x sample buffer at 95 °C for 10 min.
NOTE: After solubilization in sample buffer, store the samples long-term at −20 °C if required. - Prepare the resolving gel. Degas all the components except ammonium persulfate (APS) and TEMED for 15 min.
- Add APS and TEMED immediately before casting the gel. Carefully overlay the resolving gel with water. Allow the resolving gel to polymerize, ~30-45 min.
- Decant the water and prepare the stacking gel. Add the stacking gel, taking care to avoid bubbles. Insert a comb with the relevant number of wells, and allow to polymerize for 5 min. Monitor polymerization using any leftover stacking gel.
- Assemble the gel for running SDS-PAGE and add reservoir buffer to the chamber.
- Load 10 µL of each sample per well, or 8 µL of the protein ladder. Run at 180 V until the samples enter the resolving gel, and then reduce voltage to ~160 V and run until the dye front is ~0.5 cm from the edge of the plate.
NOTE: The time it takes for the samples to reach the bottom may vary between 1-1.5 h. The time may be lengthened by reducing the voltage. Never reduce the voltage to zero. Higher voltages may increase gel "smiling" and crack the plates. - Transfer the gel either to Coomassie stain (for protein staining) or to 1x transfer buffer (for western blotting). For Coomassie staining, stain overnight, and then destain, image, and dry.
- For western blot, prepare the transfer system according to the manufacturer's instructions.
- For a wet transfer, use cold 1x transfer buffer and pre-wet pads, filter paper, and nitrocellulose. For the Bio-rad Protean III system (see Table of Materials) used here, the filter paper may be cut to 10 cm x 7.5 cm. Cut the nitrocellulose to 9 cm x 6.75 cm.
NOTE: Nitrocellulose is highly flammable. Avoid open flames and other potential sources of ignition. - Lay out the transfer cassette with pads, filter paper, and nitrocellulose. Roll out air bubbles. Add the gel carefully.
- Add the filter paper and roll out air bubbles. Add the pad, close the cassette, and insert into the holder in the correct orientation (ensure nitrocellulose faces the red terminal). Add a stir bar and an ice pack to the side, and top off the reservoir with cold 1x transfer buffer. Transfer at 110 V for 90 min.
NOTE: The heat generated during the transfer may adversely affect the transfer. To ensure a good transfer, always use cold 1x transfer buffer. - Remove the nitrocellulose and stain with Ponceau solution for ~5 min. Rinse with ultrapure water. Mark the protein ladder in pencil and trim the blot as needed. Destain the blot using leftover transfer buffer.
NOTE: Ponceau may be reused many times. - Block the nitrocellulose in 25 mL of 5% BSA in 1x TBST at 4 °C, with shaking, overnight. Then, discard the blocking solution and add primary antibody (1:1,000) in 1% BSA in 1x TBST. Shake at 4 °C overnight.
NOTE: The primary antibody may be saved at −20 °C and reused several times. - Wash the nitrocellulose 3x for 10 min each in 1x TBST with shaking. Discard the wash and add 10 mL of HRP-conjugated secondary antibody (1:10,000) in 1% BSA in 1x TBST. Shake at room temperature for 1 h. Wash the nitrocellulose 3x for 10 min each in 1x TBST with shaking.
- Prepare ECL reagent immediately before imaging the nitrocellulose. Immediately before imaging, decant the TBST and add the ECL reagent to the nitrocellulose. Shake for 1 min. Image the gel.
Increased promastigote sensitivity to SLO in Tyrode's buffer compared to M199
The SLO sensitivity of L. major promastigotes was compared between different assay buffers. Wild-type, spt2–, and spt2–/+SPT2 promastigotes were challenged with SLO in serum-free M199 or Tyrode's buffer supplemented with 2 mM CaCl2 for 30 min prior to analysis on a flow cytometer. Suitable parasites for analysis were single cells identified by forward/side scatter profiles (Figure 3A,B). Data were collected using channels BL1 (488 excitation, 530/30 emission filter) as an autofluorescence control and YL1 (561 nm excitation, 585/16 emission filter) for PI. BL1 was used to identify potential autofluorescence or compensation issues (x-axis in Figure 3C–E). The use of the 561 nm line reduced bleed-through from PI into the BL1 channel. The fraction of dead cells was determined for each condition, and %Specific Lysis was determined. Wildtype and spt2–/+SPT2 promastigotes were resistant to SLO in serum-free M199 but had minor (<20%) specific lysis in Tyrode's buffer (Figure 4A). The sphingolipid-deficient spt2– promastigotes were sensitive to SLO in both serum-free M199 and Tyrode's buffer (Figure 4). To quantitate the increase in sensitivity from Tyrode's buffer compared to serum-free M199, the dose-response curve was fit to a logistic model, and the LC50 was calculated for each condition (Figure 4B). The spt2– promastigotes were approximately eight-fold more sensitive to SLO in Tyrode's buffer than in M199 (Figure 4B). These data demonstrate that toxin sensitivity may vary based on the buffer used. Curve fitting enables the comparison of changes in the entire dose-response curve. Thus, flow cytometry enables a medium-throughput assay to compare the toxin sensitivity of L. major promastigotes under different conditions.
MEK pathway activation in L. major independent of toxin challenge
The single-cell data on cellular survival from the cytotoxicity assay enable downstream assays at sublytic toxin doses. For example, L. major can be assayed for biochemical pathways by western blotting after toxin challenge. The MEK pathway is activated during membrane repair in mammalian cells6. It is unknown if this pathway is activated following SLO challenge in L. major or if the commercially available antibodies recognize any L. major MAPK homologs. WT, spt2–, and spt2–/+SPT2 promastigotes were challenged with a sublytic (500 HU/mL) dose of SLO and analyzed by western blotting. A ~120 kDa band was observed for phospho-MEK in the L. major promastigotes (Figure 5). For total MEK, bands at ~120 kDa and ~55 kDa were observed (Figure 5), which are consistent with the sizes of MRK1 and LmxMKK, respectively. Phospho-ERK detected similar bands, while the ERK antibody staining was not robust in this assay (Figure 5). Lipophosphoglycan (LPG) and tubulin were also blotted as loading controls. These data reveal the importance of validating antibodies for use in L. major.
Figure 1: 96-well plates enable an ordered setup for cytotoxicity assays. The cytotoxicity assay is divided into six columns of two technical replicates each. With controls, this permits two conditions to be tested per plate. In this example, L.major promastigotes resuspended in serum-free M199 are compared to those in Tyrode's buffer. The genotypes follow the order of wild-type (WT) (green), spt2– (red), and spt2–/+SPT2 (blue). The toxin (SLO) is added such that the concentration runs from the highest concentration at the top of the plate to the bottom, leaving the final row as a no-toxin control. Please click here to view a larger version of this figure.
Figure 2: Consistent setup enables reproducible cytotoxicity results. After calculating the amount of toxin needed, the volume of assay buffer necessary for a 2x solution is prepared. The viability dye (PI) and CaCl2 are added as needed and the buffer dispensed in 1.5 mL tubes. After washing and resuspending in assay buffer, equal numbers of L. major promastigotes are dispensed in each well of the 96-well plate. The toxin is added to the assay buffer and serially diluted from the highest toxin dose (1st tube) down to the lowest dose (7th tube) immediately before addition to the cells. The toxin is added to cells in the 96-well plate beginning from the bottom of the plate at the lowest toxin dose (7th row) and working up to the highest dose (1st row). Please click here to view a larger version of this figure.
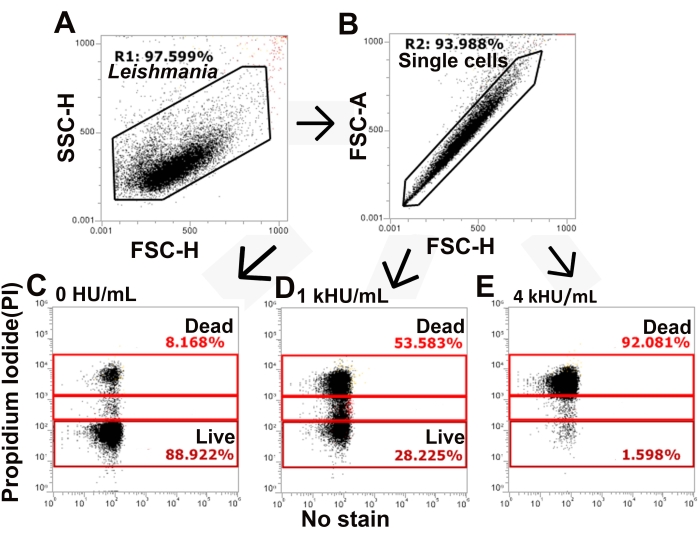
Figure 3: The gating strategy distinguishes live and dead populations of cells. L. major promastigotes stained with PI and challenged with SLO were analyzed by flow cytometry. (A,B) The gating strategy identifies single cells from total L. major based on forward and side scatter. (C–E) PI staining is next assessed, and cells are gated into PI high, PI low, and PI negative populations. Representative data for (C) no toxin, (D) 1,000 HU/mL, and (E) 4,000 HU/mL SLO are shown. Please click here to view a larger version of this figure.
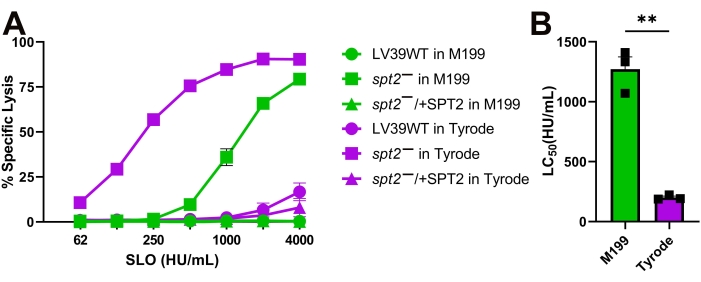
Figure 4: Tyrode's buffer enhances the sensitivity of L. major promastigotes compared to M199. Wildtype (WT), spt2–, and spt2–/+SPT2 L. major promastigotes in serum-free M199 or Tyrode's buffer supplemented with 2 mM CaCl2 were challenged with SLO at the indicated concentrations at 37 °C for 30 min, and PI uptake was analyzed by flow cytometry. The %Specific Lysis was calculated. A logistic model was constructed and the LC50 calculated for the spt2– mutants. Graphs display (A) mean ± SEM or (B) individual experiments from three independent experiments. A Student's unpaired t-test with Welch's correction was used for the LC50 values. ** p < 0.01 Please click here to view a larger version of this figure.
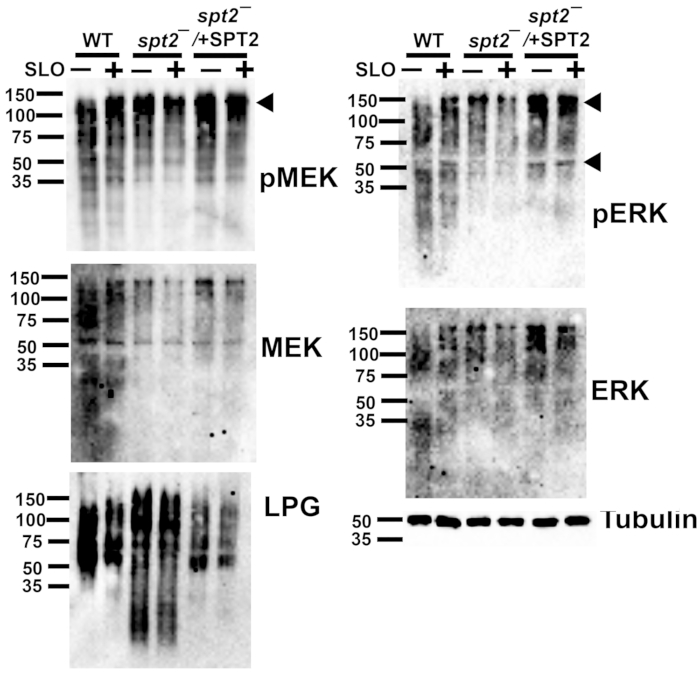
Figure 5: Phospho-ERK and phospho-MEK1/2 detect L. major proteins independently of sphingolipids or toxin challenge. Wildtype (WT), spt2–, and spt2–/+SPT2 L. major promastigotes were challenged for 30 min at 37 °C with or without 500 HU/mL SLO in serum-free M199 supplemented with 2 mM CaCl2. The cell lysates were resolved by SDS-PAGE, transferred to nitrocellulose, and probed for phospho-MEK (pMEK), total MEK, phospho-ERK (pERK), total ERK, lipophosphoglycan (LPG), or tubulin, followed by HRP-conjugated secondary antibodies and ECL. A representative blot from two independent experiments is shown in each case. Please click here to view a larger version of this figure.
Table 1: Buffer and media compositions. Please click here to download this Table.
Table 2: Sample layout for Excel calculations. Please click here to download this Table.
Supplemental File 1: Excel spreadsheet with formulae. Please click here to download this File.
