The concept and timing of this proExM protocol is depicted in Figure 1. On day 5, transfected cells are fixed and stained with fluorescent antibodies targeting the protein of interest (Figure 1A,B). On day 6, treatment with AcX leads to formation of amine groups on all proteins (including fluorophores) (Figure 1A,B)12. Upon polymerization of the hydrogel, these amine groups bind covalently to the hydrogel (day 6). After polymerization of the gel, homogenization (digestion) is performed with proteinase K resulting in the destruction of structural proteins of the cell (day 6, Figure 1A,B). Fluorescently labeled antibodies remain mostly preserved after digestion. Due to the disruption of structural proteins, water dialysis of the hydrogel results in isotropic expansion of the cell within the hydrogel on day 7 (Figure 1A,B). Imaging of the sample is performed with a conventional fluorescence microscope (Figure 1A). Data validation to determine the expansion factor and to exclude distortions should be performed (Figure 1A).
To perform expansion of the cell isotropically, the gelation step is essential. Figure 2 shows the lateral and top view of a gelation chamber. Glass cover slips build the spacers of the gelation chamber (Figure 2A1-3/C1-3). The cover glass with the fixed and stained cells is positioned with the cells upward onto a glass slide (Figure 2A4-C4). The lid of the gelation chamber is wrapped with parafilm and is closed bubble-free (Figure 2A5-C5).
This ExM protocol enables expansion of up to four-fold. To determine the expansion factor, it is essential to image cells before and after expansion (Figure 3A + B). Insufficient anchoring and homogenization may lead to distortions and ruptures of cells. Figure 4A + B shows representative examples of ruptured cells in different magnification images.
This method can be used to investigate the co-localization of F-actin and actin adaptor proteins, e.g., podocin and nephrin (Figure 5). Podocin is depicted in green while actin is labeled in blue (Figure 5). Nephrin is marked in green. White areas indicate co-localization.
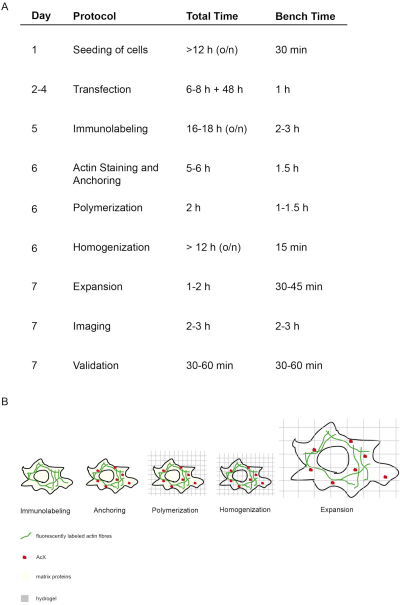
Figure 1: Concept and timing of this ExM protocol. (A) In the "protocol" column, each step of the protocol is outlined. (A + B) After seeding and transfecting cells, immunofluorescent labeling is performed (Immunolabeling). (A + B) The small molecule AcX (red dot) binds to all proteins and anchors them to the hydrogel (Anchoring). (A + B) Via polymerization all proteins including fluorophores are covalently bound via AcX to the hydrogel (Polymerization). (A + B) Homogenization leads to digestion of structural matrix proteins. (A + B) Expansion is achieved by dialysis in water. (A) Imaging and validation of imaging finalizes the experiment. (A) The entire protocol requires 7 days (column "day") with many incubation steps (total time per day column "total time"), but actual bench time is much less as indicated in the respective column "bench time". Modified from14. Please click here to view a larger version of this figure.
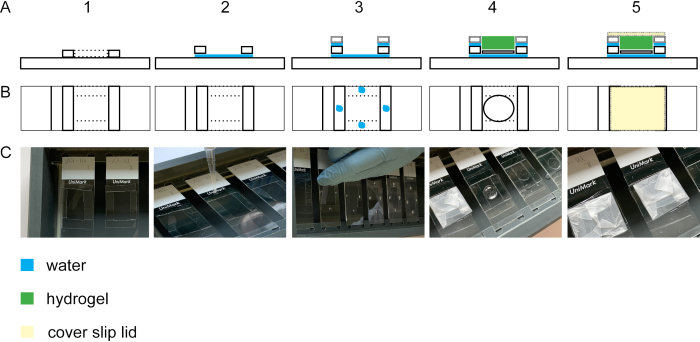
Figure 2: Building the gelation chamber. Side view (A1) and top view (B1 + C1) of a glass slide with four #1.5 cover stripes. By adding a droplet of water between the glass slide and the cover slip stripes, the stripes will adhere to the glass slide (side view A2, top view B2, C2). Droplets of water on the #1.5 cover stripes lead to adhesion of #1.0 cover stripes laid on top of the #1.5 cover stripes (side view A3, top view B3, C3). The sample on the cover slip is placed in the middle of the rectangle using forceps. The gel is pipetted on top (side view A4, top view B4, C4). (A5) Side view and top view (B5 and C5) of the assembled gelation chamber including the closed lid which is built from a cover slip wrapped in parafilm. Please click here to view a larger version of this figure.
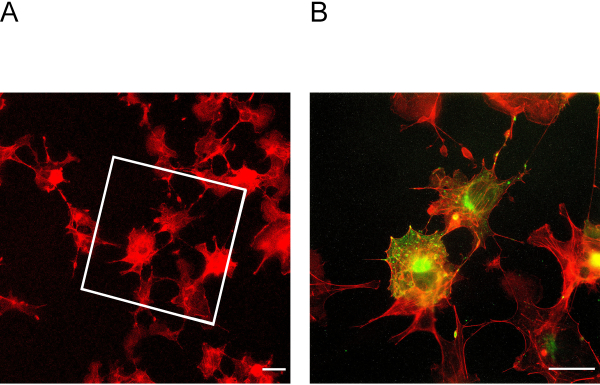
Figure 3: Cells before and after expansion. (A) Cells before expansion stained for actin. The box indicates in which area the expanded cells in Figure 3B lie. (B) Cells after expansion stained for actin in red and podocin in green. Podocin co-localizes with actin in the cell periphery. Scale bar = 5 µm, expansion factor = 2. Please click here to view a larger version of this figure.

Figure 4: Distortions and ruptures of cells. (A + B) Representative microscopic images of cos7 cells immuno-stained for actin (red). Cells were fixed, stained, anchored, digested and expanded. (A) Ruptures of cells. Arrows indicated ruptured areas. Scale bar = 5 µm, expansion factor = 4. (B) Ruptures and distortion of cells. White arrows indicated ruptured areas. Scale bar = 5 µm, expansion factor = 4. Please click here to view a larger version of this figure.
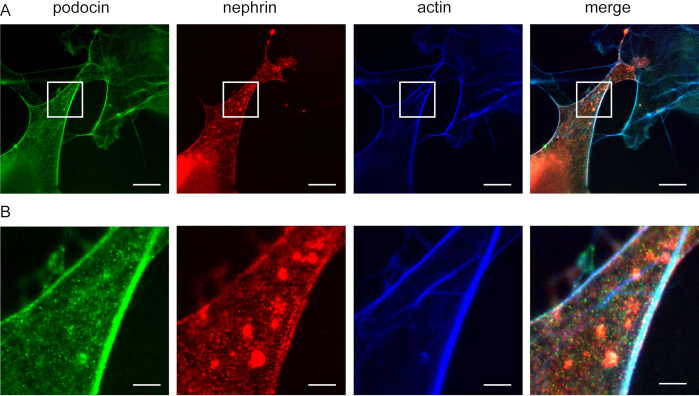
Figure 5: Podocin co-localizes with nephrin and actin. Cos7 cells immunofluorescently labeled for podocin, actin, and nephrin. (A) Cos7 cells stained for podocin (green), actin (blue), and nephrin (red) with ExM. Podocin co-localizes with actin and nephrin. Scale bar = 200 nm, expansion factor = 4. (B) Magnification of the indicated area in (A), Scale bar = 40 nm. Please click here to view a larger version of this figure.
| Solutions for ExM | |||
| Anchoring buffer | final concentration | ||
| NaHCO3 | 150 mM | ||
| Acryloyl-X, SE (AcX) | 0.1 mg/ml | ||
| Monomer solution | Stock solution concentration g/100ml | amount (ml) | final concentration (g/100 ml) |
| sodium acrylate | 38 | 2.25 | 8.6 |
| acrylamide | 50 | 0.5 | 2.5 |
| N,N’-Methylenebisacrylamide | 2 | 0.5 | 0.10 |
| sodium chloride | 29.2 | 4 | 11.7 |
| PBS | 10x | 1 | 1x |
| water | 1.15 | ||
| total | 9.4 | ||
| Gelling solution | Stock solution concentration | amount (µl) | final concentration (mg/ml) |
| monomer solution | NA | 190 | NA |
| APS | 10% | 4 | 2 |
| TEMED | 10% | 4 | 2 |
| water | NA | 2 | NA |
| total | 200 | ||
| Digestion solution | Stock solution concentration | amount (µl) | final concentration |
| Tris Cl, pH 8.0 | 1 M | 1000 | 50 mM |
| EDTA pH 8.0 | 0.5 M | 40 | 1 mM |
| Triton X-100 | 10% | 1000 | 0.5 % |
| Guanidin HCL | 8M | 2000 | 0.8 M |
| water | ad 20 ml | ||
| proteinase K | 800 U/ml | 100 | 4 U/ml |
Table 1: Solutions for ExM.
