1. Preparations
NOTE: The protocol described below has been optimized for the use with REF52 cells and ATM+/+ or ATM-/- human fibroblasts. Other cell types may require further optimization as described in the notes and troubleshooting sections below.
- Make 500 mL of complete cell culture medium for REF52 cells. To 500 mL of high-glucose containing Dulbecco’s modified Eagle’s medium (DMEM) add 10% FBS, 2 mM L-glutamine, and 100 units/mL penicillin-streptomycin.
- Prepare a 25 μg/mL solution of fibronectin (FN) by adding 300 μL of 1 mg/mL FN solution to 12 mL of sterile 1x phosphate buffered saline (PBS), pH 7.4. Mix well.
- Prepare a 0.5% (w/v) delipidated (i.e., fatty acid free) bovine serum albumin (dlBSA) solution in serum free DMEM cell culture medium. Add 0.5 g of dlBSA to 100 mL of serum free DMEM medium. Mix the solution well, but do not vortex. Sterile filter the solution into a new sterile container, using a 0.22 μM syringe filter before use. Store at 4 °C.
- Make 3.7% paraformaldehyde solution by dissolving 3.7 g of paraformaldehyde in 100 mL of 1x PBS. Use gentle heat and stirring to get the paraformaldehyde into solution.
NOTE: The paraformaldehyde solution is light sensitive and should be protected from light. It is good for up to one week when stored at 4 °C.
CAUTION: Paraformaldehyde is toxic, flammable, corrosive and a health hazard. Review the material safety data sheet for paraformaldehyde prior to use. Use the appropriate personal protective equipment when handling including eye shield, face shield, full-face particle respirator, gloves, and lab coat. - Prepare the permeabilization solution containing 0.2% non-ionic surfactant in 1x PBS (v/v). For 100 mL, slowly add 0.2 mL of Triton X-100 to 100 mL of 1x PBS, while stirring.
- Make the immunofluorescence blocking buffer containing 2.5% BSA, 5% goat serum, and 0.05% non-ionic surfactant (w/v/v) dissolved in 1x PBS solution. For 100 mL, add 5 mL of goat serum, 2.5 g of BSA and 0.05 mL of Triton X-100 in ~ 95 mL 1x PBS, while stirring.
- Grow REF52 cells in DMEM complete culture medium in a 10 cm2 (or any other vessel size) cell culture-treated plate in a cell culture incubator at 37 °C and 5% CO2.
2. Coating cell culture plates with the extracellular matrix protein fibronectin
NOTE: Perform this section using aseptic technique and sterile reagents in a BSL-2 certified laminar flow hood. Refer to Figure 1A for an overview of key steps prior to beginning.
- Using a tissue culture certified 24-well plate, place one glass coverslip (12-Cir-1) in each well. Label the plate according to Figure 1B.
- Pipette 500 μL of the 25 μg/mL FN solution to each well of a 24-well plate.
- Pipette the solution over each coverslip a few times to ensure even coating and complete submersion. Place the lid back on the plate.
- Incubate the plate in a cell culture incubator at 37 °C and 5% CO2 for 1 h.
NOTE: Alternatively, incubate overnight at 4 °C. - After 1 h, remove the plate from the incubator and aspirate the FN solution from the wells.
- Wash wells three times with 500 μL of 1x PBS. Aspirate the final wash of 1x PBS.
- Block wells with 500 μL of 0.5% dlBSA solution for a minimum of 15 min at 37 °C and 5% CO2.
- Aspirate the dlBSA solution prior to plating cells in step 3 below.
NOTE: If storing plates, add 500 μL of 1x PBS to each coverslip after aspiration of the dlBSA solution. Plates can then be kept at 4 °C for up to one week.
3. Preparing anchorage-dependent cells for the adhesion assay
NOTE: Perform this section using aseptic technique and sterile reagents in a BSL-2 certified laminar flow hood.
- At least 30 min prior to the cell plating, pre-warm the following solutions: DMEM complete medium, dlBSA solution, 1x PBS, 0.5% trypsin-EDTA solution, and trypsin neutralizing serum (TNS) in a 37°C water bath.
- Starting with a confluent monolayer of REF52 cells in a 10 cm2 dish, wash cells twice with 6 mL of warm 1x PBS. Serum starve the cells for at least 1 h (depending on cell type) in 6 mL of warm dlBSA solution at 37 °C and 5% CO2.
- Wash cells with 6 mL of warmed 1x PBS, aspirate PBS and add 1.5 mL of warm 0.5% Trypsin-EDTA solution.
- Place cells in a cell culture incubator at 37 °C and 5% CO2 for ~2 min.
- Observe cells under a light microscope to ensure the detachment is complete. If cells are still adherent after tapping the plate on the bench top, return to the 37 °C incubator for an additional 2 min. Trypsinize the cells for as little time as is necessary.
- Pipette 1.5 mL of warm trypsin neutralizing solution (TNS) to the dish to stop trypsinization and collect detached cells. Pipette the solution up and down over the bottom of the plate numerous times to remove all remaining adherent cells. If cells appear clumpy, further triturate the cell suspension by gently pipetting up and down over the back of the dish.
- Count cells using trypan blue exclusion and a hemocytometer under a light microscope. Alternatively, use an automated cell counter.
- Remove an appropriate amount of cells to create a 1.0 – 3.0 x 104 cells/mL cell suspension in 5 mL of dlBSA in a 15 mL conical tube.
- Centrifuge cells at ~ 300 x g for 5 min using a fixed angle rotor in a table-top centrifuge.
- Aspirate the supernatant from the cell pellet, and resuspend cells in a total of 7 mL of warm dlBSA solution. Do not allow the cells to be overly confluent upon coverslip plating, but evenly distributed with few cells touching one another.
- Evenly divide the cell suspension into two 15 mL conical tubes, one for the vehicle alone control (DMSO) and one for Ku55933 (ATM kinase inhibitor, oxidant)5. Ensure each tube contains 3.5 mL of the cell suspension.
- Using a tube rotator, revolve the tubes at 37 °C for 90-120 min in a cell culture incubator.
- 30 min before plating, add a final concentration of 10 μM Ku55933 and DMSO (1:1,000) to each respective tube. Place the cell suspension back on the rotator for the remaining time.
- Immediately prior to plating the cells, retrieve the 24-well plate from the incubator and aspirate the dlBSA solution.
- After revolving the cell suspension for 90-120 min, remove 500 μL of cell suspension from each treatment group and add to one FN coated coverslip in the 24-well plate from step 2 as illustrated (Figure 1B). Return the plate to the 37 °C and 5% CO2 cell culture incubator and the cell suspension back to the rotation.
- After plating the cell suspension on the FN covered-coverslips, allow cells to adhere for the desired length of time (e.g., 10 min, 15 min, 20 min, 30 min) and then immediately proceed to step 4.
4. Cell fixation and antibody staining for immunofluorescence
NOTE: The following steps are performed under non-sterile conditions and at room temperature unless otherwise stated.
- After the desired time for adhesion has passed, aspirate the cell solution from each coverslip in the plate.
- Using the sides of the well, gently dispense 500 μL of 3.7% paraformaldehyde solution onto each coverslip and wait 10-15 min.
- Remove the paraformaldehyde solution and wash each coverslip with 500 μL of 1x PBS for a total of two times.
NOTE: Dispose of paraformaldehyde waste responsibly, according to the institution’s environmental health and safety plan. - Aspirate the PBS, and permeabilize cells on each coverslip with 500 μL of 0.2% Triton X-100 in 1x PBS (v/v) for 10-15 min at room temperature.
- Wash each coverslip with 500 μL of 1x PBS three times.
- Block cells on each coverslip with 500 μL of immunofluorescence blocking buffer containing 5% goat serum, 2.5% BSA and 0.05% Triton X-100 dissolved in a 1 x PBS solution for 30-60 minutes.
- Dilute the primary anti-paxillin antibody (1:250) in the blocking buffer. Mix well and add 200 μL of the antibody solution to each coverslip. Incubate at room temperature for at least 1 h.
NOTE: Alternatively, the primary antibody solution can be incubated overnight at 4 °C. There are many common focal adhesion markers that could be used for staining adhesion complexes and subsequent FA analysis. These include antibodies against the following proteins: integrin subunits (β1, α5, or αV), talin, or vinculin2. - Aspirate the antibody solution, and wash each coverslip with 500 μL of 1x PBS three times for 10 min each. Protect the samples from light from this point forward.
- Dilute the phalloidin F-actin probe conjugated to the red fluorescent Alexa 594 dye (1:1000) and goat-anti mouse 488 fluorescent secondary antibody (1:400) in the same blocking buffer solution. Mix well and add 200 μL of the antibody solution to each coverslip for 30 min.
NOTE: Fluorescently conjugated secondary antibodies from other species may be used as well. However, the use of antibodies from other species will require modification of the blocking buffer serum. - Aspirate the antibody solution, and wash each coverslip with 500 μL of 1x PBS three times for 10 min each.
- Aspirate the 1x PBS and rinse one time with 500 μL of dIH2O.
- Mount coverslips onto microscope slides using anti-fade mounting medium containing DAPI.
- Leave microscope slides to set overnight in the dark at room temperature.
- Store microscope slides in the dark at 4 ˚C for the long-term storage and until imaging.
NOTE: Image using standard immunofluorescence techniques. It is recommended to use a high-powered oil immersion 60x objective lens to ensure enough resolution to note the focal adhesions and peripheral ruffles at cell edges. Acquire images of 20-30 cells in multiple fields of view for each coverslip under each treatment condition and time. From combined replicates, this should yield at least 60 cells in order to perform statistical analysis. Save and export fluorescence images as a .TIFF file with a minimum of 300 dpi resolution.
5. Quantifying stress fibers, cell circularity, and focal adhesion formation
NOTE: The following image analyses are performed using the latest version of the open source imaging processing package Fiji Is Just Image J (Fiji), which can be downloaded free of charge at (http://fiji.sc/).
- General image processing
NOTE: All images will need to be prepared for computational analyses by performing steps 5.1.1-5.1.5 below (Figure 2). Afterward, any or all subsequent quantification procedures may be selected.- Open the .TIFF fluorescence image using Fiji. Ensure the images are 8-bit and grayscale.
- Select Image-Adjust-Window/level and select Auto (Figure 2A).
- Select Process-Subtract Background to subtract the background fluorescence. Check Sliding Paraboloid and select the option of a Rolling Ball Radius of 50 pixels (Figure 2B).
NOTE: To verify the proper size for the rolling ball radius, select the Line Tool and draw a radius on the largest adhesion in the image. Select Measure to verify the length of the line drawn. If the value of the radius is too large, features including adhesions will be lost in the image. If the radius is too small, it will give rise to artifacts in the processed image due to background noise. - Select Image-Adjust- Brightness/Contrast to check the intensity of the adhesion over the background. Adjust if necessary.
NOTE: To optimize the brightness/contrast and avoid saturating the signal, use the lookup tool of the image to examine its histogram to adjust the brightness/contrast. - Select the following parameters under Analyze-Set Measurements: Area, Mean Gray Value, Shape Descriptors, and Integrated Density.
- Stress fiber formation analysis
NOTE: Stress fibers can be quantified multiple ways depending on the phenotype.- Count the number of cells with stress fibers as a percentage over the total number of cells. This analysis is best if there are visual differences in the number of stress fibers formed under different experimental conditions.
- Count the number of stress fibers that transverse the cell. This analysis allows for the comparison of the number of stress fibers formed per cell.
- Measure the total fluorescence intensity given by the phalloidin (e.g., F-actin) staining per cell17,18. This method will highlight drastic increases/decreases in fluorescence intensity due to F-actin staining.
- Set the measurement parameters in step 5.1.5 above.
- Select the Freehand Tool in the Fiji toolbar and manually trace the cell(s) of interest. Select Analyze-Measure. A new window will appear showing the selected measurement parameters.
- Select the Freehand Tool in the Fiji toolbar and manually trace an empty space with no cells present. Select Analyze-Measure. This measurement will serve as the background fluorescence.
- Use the equation below to determine the total F-actin fluorescence per cell:
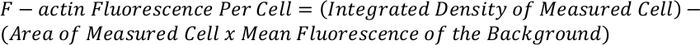
NOTE: The resulting measurement can be normalized and compared to other cells to give F-actin fluorescence per cell.
- Cell circularity analysis
NOTE: Information on cell area (an indicator of cell spreading over time), as well as, the circularity can also be recorded. This measurement is given as a ratio between 0 to 1 as a way to quantify cells that are elongated to round, respectively.- Select the Freehand Tool in the Fiji toolbar, and trace an individual cell. Select Image-Measure and record the cell area and perimeter measurements for each cell. Repeat this procedure for each cell.
NOTE: Under the Set Measurements function, circularity is provided as the Shape Descriptors measurement (step 5.1.5). - Manually count actin-enriched ruffling or protrusions per cell as depicted in Figure 3 and Figure 4.
- Select the Freehand Tool in the Fiji toolbar, and trace an individual cell. Select Image-Measure and record the cell area and perimeter measurements for each cell. Repeat this procedure for each cell.
- Focal adhesion analysis
NOTE: Before performing focal adhesion analysis, install the Mexican Hat Filter plugin on the latest version of Fiji. The following protocol has been modified from previous studies19,20,21.- Select Process-Enhance Local Contrast (Clahe) using a block size of 19, histogram bins 256, and a maximum slope of 6, with no mask and not fast. (Figure 2C)
- Select Process-Filters- Gaussian Blur with a Sigma (Radius) of 2.0 to filter the image (Figure 2D).
- Select Plugins-Mexican Hat Filter (Mhf) with a Radius of 2.0 (Figure 2E).
- Run Threshold and select Dark Background and Over/Under using either Huang or Isodata as the thresholding method. Select Auto-Threshold.
NOTE: This step ensures that adhesions are highlighted, but also distinct from one another. - Select Analyze-Analyze Particles with the following parameters selected: size=20, pixels-infinity and circularity=0.00-0.99. Check the outlines to ensure the proper detection and separation of focal adhesions.
NOTE: These results yield the number, area, and shape description of individual focal adhesions.
A general schema of the experimental set-up
Figure 1 represents the general schema for the cell adhesion and spreading protocol beginning with serum starvation of REF52 cells and ending with computational analysis of acquired fluorescence images. Key steps in the protocol are illustrated in the timeline. Of note, step 2 of the protocol describes the preparation of the FN-coated coverslips, which should be performed concurrently with step 3: serum starving REF52 cells prior to placing them in suspension (Figure 1A). An example of a mock-labeled 24-well plate indicating treatment groups and duration of cell adhesion prior to fixation of samples for fluorescence microscopy (Figure 1B).
Immunofluorescence image processing for focal adhesion quantification
REF52 cells were held in suspension for 90 minutes, plated on FN, and allowed to adhere for an additional 15 minutes. After fixation and staining with an anti-paxillin antibody, fluorescent 8-bit grayscale images of the cells were acquired. Image processing analysis was performed according to the protocol delineated in Step 5. Shown are representative images of each distinct processing step including the original image (Figure 2A), and images following background subtraction (post-Rolling Ball) (Figure 2B), CLAHE (Figure 2C), Gaussian Blur (Figure 2D) and Mexican Hat Filter (Post-MHF) (Figure 2E) filtering steps. After completing all image processing steps, individual focal adhesions should be prominent, in focus, and readily distinguishable from one another (Figure 2E). After the images are filtered, the focal adhesions can be quantified and their area measured (steps 5.1 and 5.4).
Visualization of cell adhesion and spreading on FN after oxidative stress
A representative grayscale fluorescence image of anti-paxillin (focal adhesion marker) (Figure 3, top panel) and phalloidin F-actin probe staining (Figure 3, bottom panel) of REF52 cells after plating on FN with or without Ku55933 (ROS-inducing agent) treatment. Prior to the assay, REF52 cells were serum starved for 1 h. Following serum starvation, cells were held in suspension while being treated with either vehicle alone or 10 μM Ku55933 to induce oxidative stress. Cells were plated on FN-coated coverslips for the indicated times, fixed, and then stained with an antibody to focal adhesions and phalloidin to detect F-actin proteins. Prominent focal adhesions and stress fibers should be readily visible in REF52 cells after being allowed to adhere for 20-30 min on FN. Plated cells should not overlap with one another to permit full cellular spreading after adhesion. Notice the clear, distinct cell edges as well as space for individual cells to spread (Figure 3). F-actin enriched ruffles at the leading edge of cell membranes are visible and indicated with an arrow (Figure 3, bottom panel).
Graphical representation of quantified fluorescence images of stress fibers and the degree of cell spreading
Examples of quantified images displayed in bar graph form representing the percentage of cells with stress fibers and the degree of cell spreading with and without Ku55933 treatment at various times after adhesion. Fluorescent images of phalloidin F-actin probe and anti-paxillin staining, similar to images shown in Figure 3, were analyzed for the percentage of stress fibers and cell spreading (i.e., circularity index) using the image analysis procedures described in step 5 of the protocol. Notably, oxidant treatment caused a significant increase in stress fiber formation at all adhesion time points examined (Figure 4A) and a decrease in cell spreading following 15 minutes of cell adhesion to FN (Figure 4B).
Non-quantifiable immunofluorescence images due to cellular over confluency
Serum-starved REF52 cells were held in suspension for 90 minutes, during which time they were treated with 10 μM Ku55933 to induce ROS formation. Cells were then plated on FN and allowed to adhere for 20 minutes, after which they were fixed and stained with anti-paxillin or phalloidin-Alexa 594 F-actin probe. Plating at higher cell densities leads to cellular crowding, which prohibits cells from fully spreading due to over confluency. Notice cell edges are indistinguishable from adjacent cells (yellow arrows) (Figure 5A). As a result, quantification of individual cells is precluded, and spreading circumference cannot be accurately determined. In Figure 5B, a separate cell line, mouse embryonic fibroblasts (MEF), were held in suspension and then plated on FN for 30 minutes. Cells were then fixed and stained with an anti-paxillin antibody. Out of focus cells are denoted by red arrows (Figure 5B). Furthermore, the cross-reactivity of the anti-paxillin antibody with cellular debris (blue arrow) will alter thresholding during quantitative image analysis (Part 5) and should not be included in the analysis (Figure 5B).
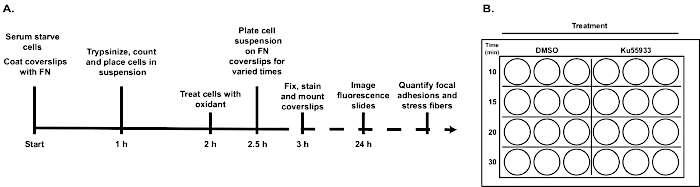
Figure 1: Time-line of protocol and example 24-well plate set-up.
(A) The time-line highlights key steps in the cell adhesion and spreading procedure. (B) Representative labeled 24-plate, illustrating treatment groups and times for cell adhesion. Please click here to view a larger version of this figure.
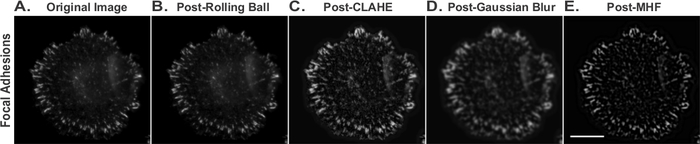
Figure 2: Examples of representative immunofluorescence images following image-processing.
REF52 cells were held in suspension for 90 min, plated on FN, and allowed to adhere for 15 min. Cells were fixed and stained with an anti-paxillin antibody. (A) Original image and images following (B) Background subtraction (post-Rolling Ball), (C) CLAHE, (D) Gaussian Blur and (E) Mexican Hat Filter (Post-MHF) filtering steps. Bar, 20 μm. Please click here to view a larger version of this figure.

Figure 3: Representative immunofluorescence images of anti-paxillin and phalloidin F-actin probe stained REF52 cells plated on FN.
Prior to the assay, REF52 cells were serum starved for 1 h. Following serum starvation, cells were held in suspension while treated with either vehicle alone or 10 μM Ku55933 to cause oxidative stress. Cells were plated on FN-coated coverslips for the indicated times, fixed and stained with an antibody to focal adhesions and phalloidin to detect F-actin proteins. F-actin enriched ruffles at the leading edge of cell membranes are indicated with an arrow. Bar, 40 μm. This figure has been modified from Tolbert et al.5 Please click here to view a larger version of this figure.
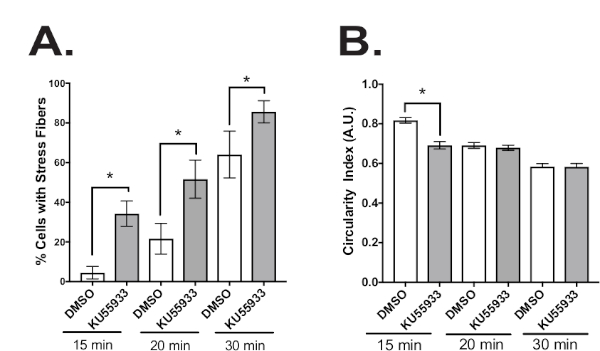
Figure 4: Quantification of immunofluorescence images.
Graphs illustrating (A) the percentage of cells exhibiting stress fibers and (B) cell circularity measurements. Cell circularity was defined as the cell area divided by the cell perimeter. Values ranged from 0-1.0 indicating an elongated or rounded morphology, respectively. Error bars indicate S.E.M. Student’s t-test for paired samples *p<0.01 from experiments performed in triplicate. This figure has been modified from Tolbert et al.5 Please click here to view a larger version of this figure.
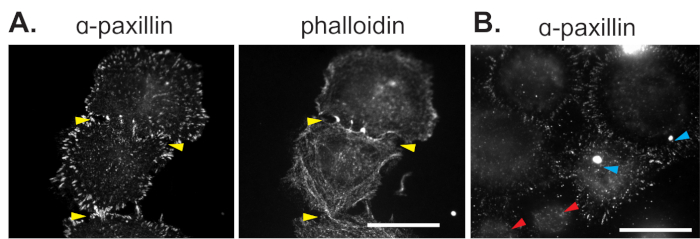
Figure 5: Non-quantifiable immunofluorescence images.
(A) Serum-starved REF52 cells were held in suspension for 90 min, while treated with 10 μM Ku55933. Cells were plated on FN and allowed to adhere for 20 min. Cells were fixed and stained with anti-paxillin or phalloidin-Alexa 594 F-actin probe. Cell edges are shown by yellow arrows. (B) MEF cells were held in suspension and then plated on FN for 30 min. Cells were fixed and stained with an anti-paxillin antibody. Out of focus cells are denoted by red arrows and cross-reactivity of anti-paxillin antibody with cellular debris are denoted with blue arrows. Bar, 30 μm. Please click here to view a larger version of this figure.
