The steps described above and described graphically in Figure 1 were applied to a human prostate tumor cell line DU145. The RNA sample was cytoplasmically prepped and was next-gen sequenced in a poly-A selected, strand-specific, paired-end protocol. Using Bowtie, the paired-end sequencing files were aligned allowing only unique matches in which the paired-end read matched better to one genomic location compared to any other genomic location. The DU145 sequence files were aligned to the human reference genome creating a bam file, which is available upon author request. Using bedtools, data was extracted from the DU145 strand-separated bam files on the number of reads that mapped to full length L1s. Those reads were sorted in a spreadsheet from largest to smallest and manually curated by examining the genomic environment around each L1 locus in IGV to confirm its authenticity (Supplemental Table 1). If a sample was curated to be authentically expressed, it was color-coded green with an explanation for its acceptance in the right most column. Examples of L1 loci accepted to be authentically expressed following guidelines described in the methods section are shown in Figure 2a-b. If a sample was rejected to be authentically expressed, it was color-coded as red with the reason for rejection on the right most column. Examples of L1 loci rejected because of expression from a promoter other than their own following guidelines described in the methods section are detailed in Figure 2c-e.
Here, only full-length L1s with an intact promoter region were studied. If this distinction is not made, a large source of transcriptional noise originating from truncated L1s is introduced. Examples of truncated L1s in DU145 are shown in Figure 3a-b where they were identified as having uniquely mapped RNA-Seq reads. In IGV, however, it is apparent that those transcripts were not initiated from the truncated L1, but from the inclusion of the L1 sequence in a gene or downstream from an expressed gene.
Overall in DU145, the percentage of full-length L1 loci and reads that are rejected as authentically expressed L1s after manual curation is approximately 50% (Supplemental Table 2) demonstrating the high level of L1 mapped transcript reads that would otherwise be recorded as false positives without manual curation. Specifically, in DU145 there were 114 total full-length L1 loci to have uniquely mapped reads in the sense direction with a total of 3,152 reads, but there were only 60 loci identified to be expressed off their own promoter after manual curation with 1,879 reads (Supplemental Table 1). This is the case even when steps were taken to reduce expression irrelevant to L1 biology by selecting for cytoplasmic mRNA. Note that the locus with the highest level of mapped transcripts in DU145 was rejected because it was not an authentically expressed L1 (Figure 4). Overall the number of mapped transcripts to specific L1 loci ranges similarly between the accepted and rejected L1 loci as authentically expressed after manual curation (Figure 4).
After manual curation, the number of reads that map uniquely to authentically expressed specific L1 loci in DU145 range from 175 reads to an arbitrarily chosen minimum cut off of 10 reads (Figure 5). This approach of identifying uniquely mapped transcript reads to L1s limits the ability to accurately quantify expression. To account for this, a correction factor for each locus based on its mappability was created. To create this correction factor, first bedtools was used to extract the number of uniquely mapped reads from the HeLa genomic bam file that aligned to all full-length L1 loci and graphed those loci from highest to lowest mapped transcript reads (Supplemental Figure 1). It was arbitrarily designated that L1s with 400 reads had full coverage mappability. The number of reads able to map to a L1 locus in HeLa genomic sequencing sample was scaled relative to 400 reads and that scaled number was then multiplied to the number of reads that mapped to each authentically expressed L1 loci in DU145 (Supplemental Table 2). As expected, the L1 elements that had larger correction scores for mappability came from younger subfamilies like L1PA2 (Supplemental Table 2). Once reads were adjusted for mappability scores in each locus, the quantitation for expression for most loci increased (Figure 6). The number of reads that mapped uniquely to authentically expressed specific L1 loci with mappability corrections in DU145 ranged from 612 to 4 reads and there was a re-ordering of highest to lowest expressing loci (Figure 6).
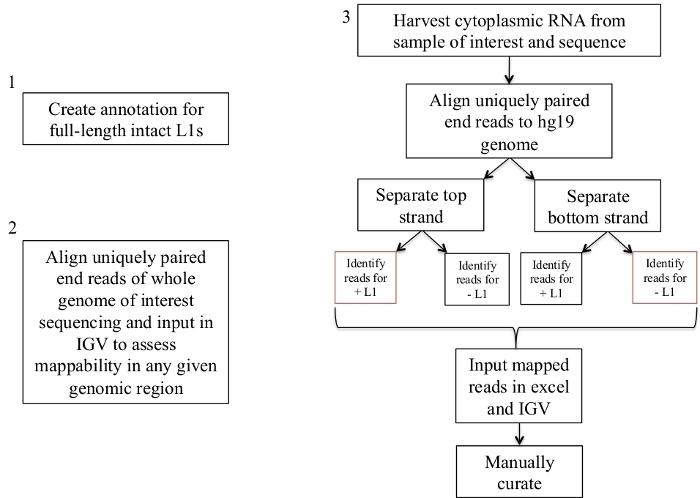
Figure 1: Workflow schematic.
Graphically described are the steps to identify expressed L1s in a human sample. Note that steps 1 and 2 do not need to be repeated if the appropriate files are already available. These appropriate files may be downloaded from Supplement File 1a-b and Supplement File 2. The boxes in red indicate the steps where bedtools coverage program is used to count the number of reads mapping to L1s in the same sense direction. These loci with sense oriented mapping reads are the L1s that should be manually curated. Please click here to view a larger version of this figure.
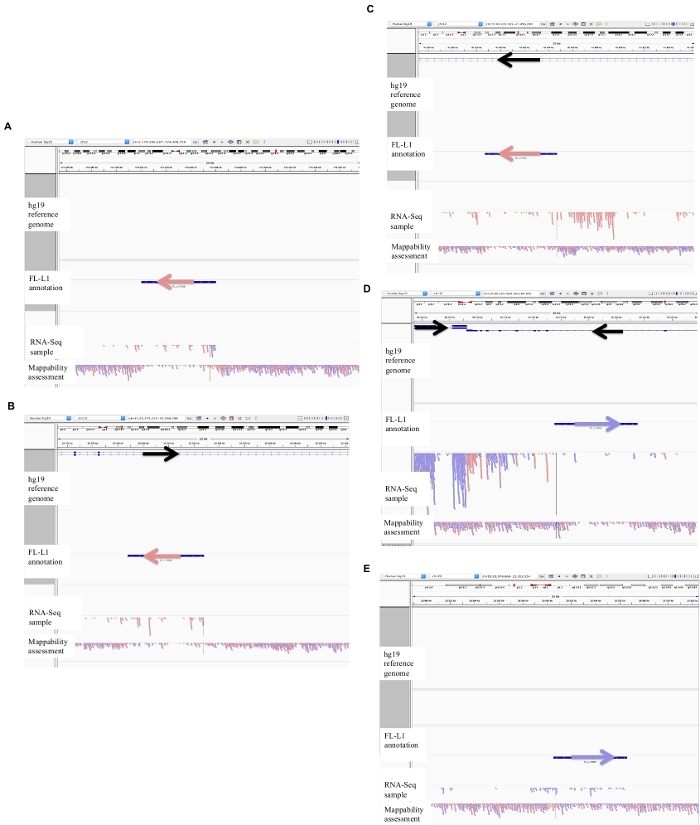
Figure 2: Examples of curated L1 loci in DU145.
Loaded into IGV are the reference genome, the full-length L1 gff annotation file matching the reference genome version (Supplement File 1), the DU145 bam file, and lastly the genomic HeLa bam file to assess mappability, which are all available upon author request. Arrows have been added to aid in the visualization of direction of the annotated L1. Arrows and reads in red are oriented in sequence from right to left. Arrows and reads in blue are oriented in sequence from left to right. a) In IGV, this L1 locus appears to be expressed off its own promoter as there are no reads upstream of the L1 in the sense orientation for over 5 kb. This L1 has low mappability, it is not in a gene, and has evidence of expected antisense promoter activity26. b) In IGV, this L1 locus appears to be expressed off its own promoter as there are no reads upstream the L1 in the sense orientation for over 5 kb. This L1 has low mappability and is within a gene of opposite direction. c) In IGV, this L1 locus was rejected as an expressed L1 as there are upstream reads in the same orientation within 5 kb. This L1 is within a gene of the same direction so the transcript reads are most likely originating from the promoter of the expressed gene. d) In IGV, this L1 locus was rejected as an expressed L1 as there are upstream reads in the same orientation within 5 kb. This L1 is downstream of a highly expressed gene in the same direction so the transcript reads are most likely originating from the promoter of that expressed gene and extending beyond the normal gene terminator. e) In IGV, this L1 locus was rejected as an expressed L1 as there are upstream reads in the same orientation within 5 kb. This L1 is not within or near an annotated gene in the reference gene so the origin of these transcripts within and upstream of the L1 element suggest an un-annotated promoter. Please click here to view a larger version of this figure.
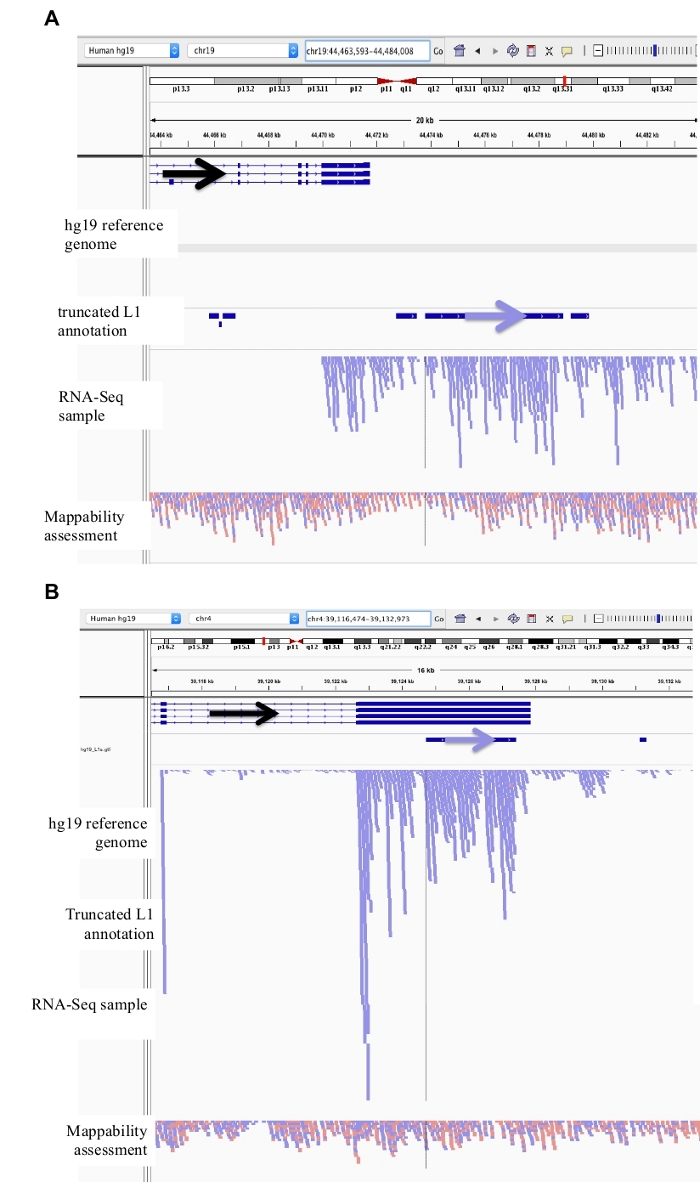
Figure 3: Background noise originates from truncated L1s as well.
Our L1 annotation does not include truncated L1s as they are a major source of background noise. Arrows have been added to aid in the visualization of direction of the annotated L1. Arrows and reads in blue are oriented in sequence from left to right. a) Demonstrated is an example of a truncated L1 in the L1MB5 sufamily that is 2706 bps. In IGV it is apparent that the reads originate from downstream extension of an expressed gene. b) Shown is another example of a truncated L1. This L1 is an L1PA11 that is 4767 bps long. In IGV it is apparent that the reads mapping uniquely to the L1 originate from the expressed exon, which the L1 is within. Please click here to view a larger version of this figure.
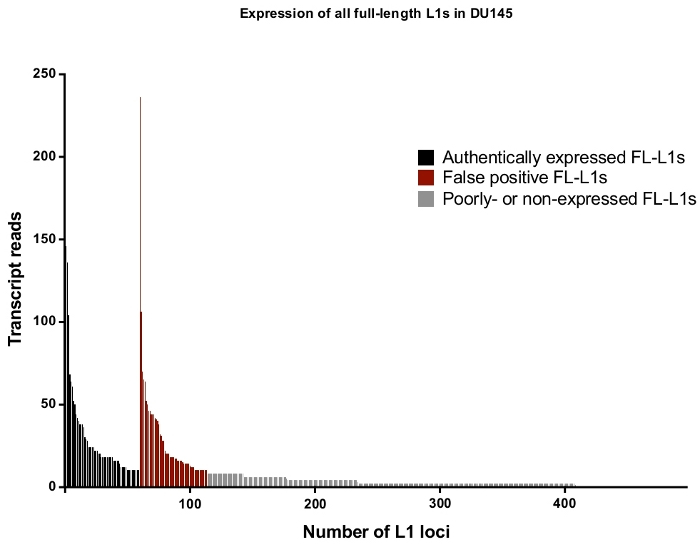
Figure 4: Transcript reads that map uniquely to all full-length intact L1s in the human genome expressed in DU145 prostate tumor cell line.
In black are the specific loci to be identified as authentically expressed after manual curation and in red are the specific loci to be rejected as authentically expressed reads after manual curation. In grey are loci with less than ten reads mapping to each. As these loci represent a small fraction of transcript reads, they were not manually curate. The x-axis tick marks denote every 100 full-length, intact L1s. Approximately 4,500 loci are not graphically shown as they had zero mapped reads. Please click here to view a larger version of this figure.
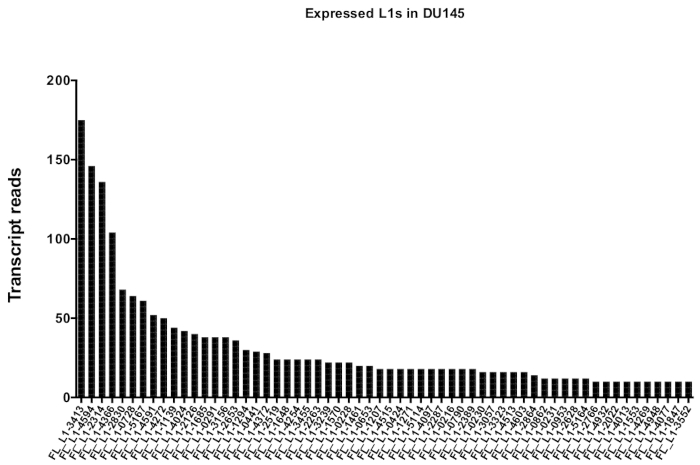
Figure 5: Transcript reads that map uniquely to authentically expressed full-length intact L1s in DU145 prostate tumor cell line.
Shown are the numbers of transcript reads that map to specific loci in DU145 cells after manual curation. Please click here to view a larger version of this figure.
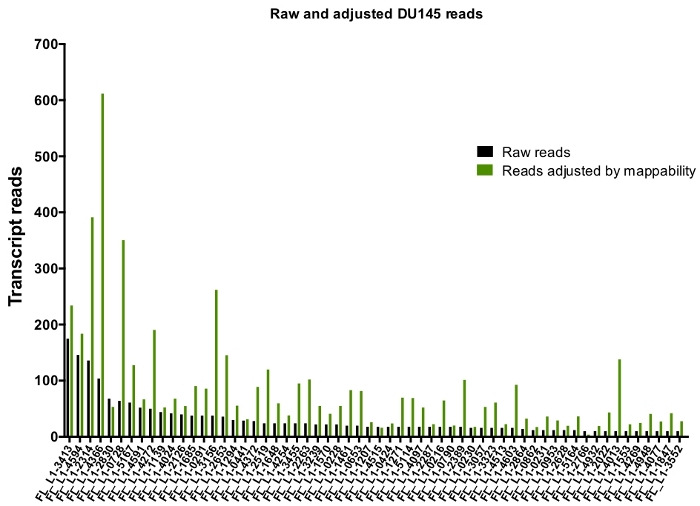
Figure 6: Reads mapping to authentically expressed L1 when adjusted by mappability.
Shown are the numbers of transcript reads adjusted by loci-specific mappability scores that map to manually curated L1 loci in DU145 cells. Please click here to view a larger version of this figure.
Supplemental File 1: Annotations for full-length, intact human L1s according to orientation. a) FL-L1-BLAST_RM_minus.gff. b) FL-L1-BLAST_RM_plus.gff. Please click here to download this file.
Supplemental File 2: Supercomputer scripts used to automate the bioinformatics pipeline detailed in section 4. Please click here to download this file.
Supplemental Figure 1: Genomic DNA sample used to determine L1 mappability.
Shown are the number of genomic transcript reads from HeLa cell line sample that map uniquely to all 5,000 full-length L1 loci in the genome. It was designated that an L1 has full coverage mappability when 400 reads map to the L1. Please click here to download this figure.
Supplemental Table 1: Manual Curation of L1s in DU145. Please click here to download this table.
Supplemental Table 2: Curated L1s in DU145 with mappability adjustment. Please click here to download this table.
