MSI-CE-MS enables the serial injection of ten or more discrete samples within a single run, which greatly enhances throughput (<3 min/sample) without complicated instrumental modifications, column-switching programs, or costly infrastructure investments (Figure 1A). An alternating series of hydrodynamic injections of the sample and electrokinetic spacer of BGE is performed within an unmodified fused-silica capillary, where zonal electrophoretic separations of ions occur under strongly acidic electrolyte conditions (pH 1.8). Solute ionization also occurs under steady-state conditions. In this case a coaxial sheath liquid, with a mass calibrant, is used as an interface for CE-MS under the positive ion mode detection with minimal ion suppression or enhancement effects as monitored by the mass calibrant ions signal. TOF represents a robust yet cost-effective HRMS with a fast data acquisition ideally suited for the nontargeted screening/drug surveillance applications when using MSI-CE-MS. For example, impressive resolution is achieved for various isobaric/isomeric DoA and their metabolites, including structural isomers of three opioid structural isomers, namely norhydrocodone, hydromorphone, and morphine (Figure 1B). In this case, 30 resolved peaks from 10 independent samples of a drug mixture are detected without sample carry-over. A synthetic urine blank/negative urine control is also included within the serial injection series (sample position #6). Also, two other isobaric opioids, namely 6-acetylmorphine (an inactive heroin metabolite) and naloxone (an opioid receptor antagonist used for the emergency treatment of opioid overdose) are fully resolved as 20 distinct peaks with full-scan data acquisition (Figure 1C). Similarly, two amphetamine positional isomers are fully resolved in MSI-CE-MS to distinguish illicit methamphetamine abuse from the potential misuse of phentermine, a prescribed stimulant that is used as an appetite suppressant for weight loss.

Figure 1: A series of extracted ion electropherograms (EIE) for representative DoA isomers/isobars that are resolved by MSI-CE-MS by analyzing ten samples and a blank within a single run. (A) Schematic of MSI-CE-MS that depicts the serial injection configuration used for an 84-DoA panel in synthetic urine. This multiplexed separation method uses an alternating series of hydrodynamic injections of 11 discrete samples and blank with an electrokinetic injection of a buffer to initiate the zonal electrophoretic separation of ions, followed by a full-scan data acquisition by TOF-MS with positive ion mode detection. (B) Three isobaric functional group opioid isomers (m/z 286.1438), separated by CE, comprising 30 resolved peaks from 10 discrete sample injections, including norhydrocodone, hydromorphone, and morphine. (C) Two isobaric opioid drugs and their metabolites (m/z 328.1543), separated by CE, comprising 20 resolved peaks from 10 discrete sample injections, including 6-acetylmorphine (heroin metabolite) and naloxone. (D) Two isobaric functional group amphetamine isomers, separated by CE, comprising 20 resolved peaks from 10 discrete sample injections, including methamphetamine and phentermine. In all cases, negative urine controls/blanks introduced at the sixth sample position in MSI-CE-MS had negligible evidence of sample carry-over. Please click here to view a larger version of this figure.
To screen DoA, a serial injection configuration in MSI-CE-MS used an 84-drug panel mixture at the recommended screening cut-off concentration levels (as the first injection position for all runs) followed by a randomized analysis of 10 representative urine samples from clinically depressed patients with a known prescription history. For instance, a positive screening test result for methadone (m/z 310.2165) from patient #208 (Figure 2A) is deduced by a detection of a large signal peak (originating from injection #9) which comigrates with methadone-d3 with a low mass error (<5 ppm). No other signals are detected in any other urine samples within the same run. Methadone concentration exceeds 13x the recommended cut-off limit when comparing its measured ion response ratio in the sample (injection #9) with the reference drug mixture/QC (injection #1) and corrected by a fourfold urine dilution factor. Thus, this result confirms the patient's adherence to methadone maintenance therapy. Evidence of nonprescribed amphetamine intake (Figure 2B) is shown by elevated amphetamine levels (m/z 136.1121) which was only detected in one patient (patient #50, in injection #11) within the MSI-CE-MS run. The measured concentration slightly exceeded the recommended cut-off levels (1.3x). This comigrates with amphetamine-d5 and had a low mass error with a top-ranked molecular formula match. A positive test result for the antidepressant venlafaxine (m/z 278.2115), a selective serotonin-norepinephrine reuptake inhibitor, is also demonstrated in patient #281 (injection #6). In this case, the concentration exceeds the recommended cut-off levels (15x), This is identified by its accurate mass- or molecular formula-match, together with comigrating venlafaxine-d6 (Figure 2C). The latter criterion is not met for an unknown isobar that is also detected within the EIE trace, which highlights the need for caution when relying solely on its accurate mass. Also, a definitive detection of prescribed pregabalin, which is prescribed for the treatment of neuropathic pain, as well as of generalized anxiety, is also demonstrated for patient #309, based on its grossly elevated concentration (64x) above the recommended cut-off levels (Figure 2D). It is, however, not detected in the nine other patient urine samples analyzed within the same run. Similar to the other screen-positive cases, injection #4, comigrates with pregabalin-d6, which is included in all urine samples analyzed by MSI-CE-MS.
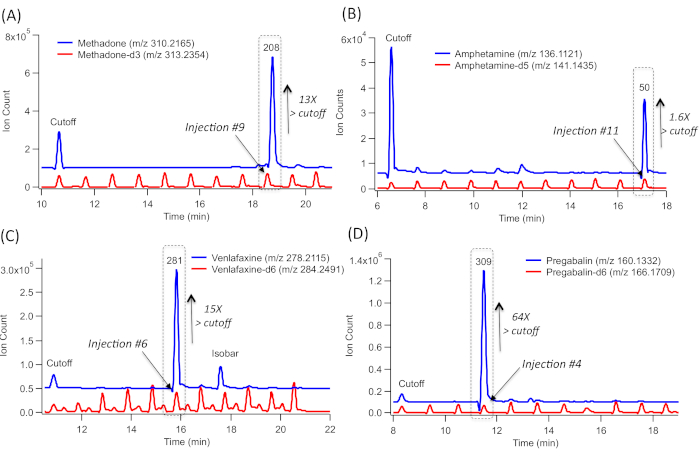
Figure 2: A series of extracted ion electropherograms (EIE) for representative screen-positive urine drug test results from a cohort of 10 clinically depressed patients as confirmed when using MSI-CE-MS, which includes an 84-drug panel at recommended cut-off level, injected as the first sample injection position serving as internal reference/QC. (A) EIE overlay corresponding to methadone-d3, which comigrates with methadone, highlighting that only one urine sample (injection position #9) has elevated methadone concentrations far exceeding the cut-off level. (B) EIE overlay corresponding to amphetamine-d3, which comigrates with amphetamine, highlighting that only one urine sample (injection position #11) has elevated concentrations above cut-off levels. (C) EIE overlay corresponding to venlafaxine-d6, which comigrates with venlafaxine, highlighting that only one urine sample (injection position #6) has elevated concentrations above cut-off levels. (D) EIE overlay corresponding to pregabalin-d6, which comigrates with pregabalin, highlighting that only one urine sample (injection position #4) has grossly elevated concentrations exceeding cut-off levels. All urine samples were analyzed directly after a fivefold dilution in deionized water together with the addition of a matching d-IS (when available). A screen-positive result in MSI-CE-MS corresponds to a drug that comigrates with d-ISs with a low mass error (<5 ppm) and the correct molecular formula, whose concentration exceeds the cut-off that is analyzed within the same run as internal reference/QC. Please click here to view a larger version of this figure.
The absolute quantification of DoA and their metabolites is also achieved by MSI-CE-MS based on external calibration curves, using reference calibrant standards for DoA that are acquired rapidly within a single run. For instance, the serial dilution of an 84-drug calibrant mixture together with a matching d-IS at a fixed concentration provides a reliable quantitative analysis in complex urine samples. This compensates for the potential matrix-induced ion suppression or enhancement but also for variations in the injection volume in the capillary between the samples. In cases when a matching d-IS is commercially unavailable or cost-prohibitive for certain DoA, a surrogate IS is used for data normalization, such as a d-IS from within the same drug class or a synthetic compound not found in urine (F-Phe), as demonstrated previously14. Representative EIEs and external calibration curves for oxycodone are shown in Figure 3A, B. and for citalopram in Figure 3C, D. These are widely prescribed analgesics and antidepressants, respectively, with abuse potential. Relative ion response ratios to their matching d-IS are measured. In this case, a serial injection configuration in MSI-CE-MS, comprising five different drug calibrants, are analyzed in duplicate within a single run together with a synthetic urine blank. Overall, good linearity (R2 > 0.990) over a 20-fold concentration range was achieved with adequate sensitivity for the detection of the majority of DoA and their metabolites (i.e., cationic alkaloids) within the 84-drug panel14. In all cases, drug metabolites are detected as their protonated molecular ion [MH+] above their screening cut-off limits (>50 ng/mL), with the exception of certain acidic/neutral drugs that have a poor ionization efficiency under the positive ion mode, such as cannabinoids (e.g., THC-COOH), barbiturates (e.g., secobarbital), and carbamates (e.g., carisoprodol).
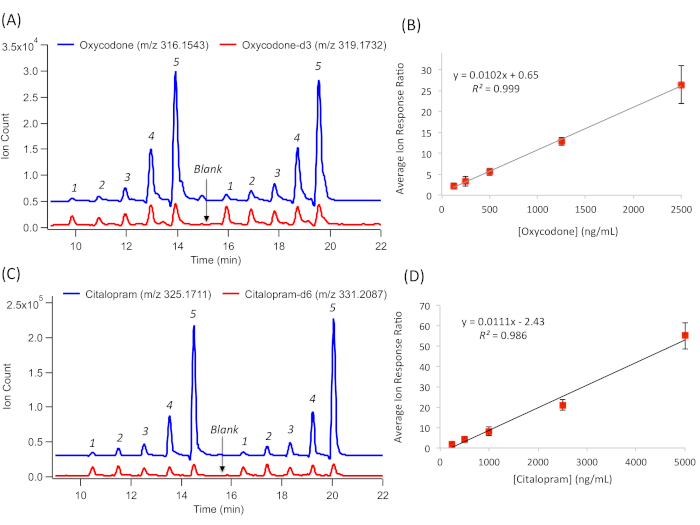
Figure 3: A series of extracted ion electropherograms (EIE) for the quantification of the representative DoA. This is done by generating external calibration curves based on their relative ion response ratio with a a matching d-IS over a 20-fold linear dynamic range. (A) Duplicate injection of a five-point calibration curve for oxycodone calibrants together with oxycodone-d3 and a blank. (B) External calibration curve for oxycodone, following linear regression to derive sensitivity (slope) and linearity (R2), with error bars representing ±1σ (n = 4). (C) Duplicate injection of a five-point calibration curve for citalopram calibrants, together with citalopram-d6 and a blank. (D) External calibration curve for citalopram, following linear regression to derive sensitivity (slope) and linearity (R2), with error bars representing ±1 SD (n = 4). Please click here to view a larger version of this figure.
