1. PGMA-b-PVDMA Synthesis20
- Synthesis of PGMA macro-chain transfer agent (Macro-CTA)
- Use a 250-mL round-bottom reaction flask equipped with a polytetrafluoroethylene-coated magnetic stir bar.
- Combine 14.2 g of glycidyl methacrylate GMA (142.18 g/mol) with 490.8 mg of 2-cyano-2-propyl dodecyl trithiocarbonate (CPDT) (346.63 g/mol), and 87.7 mg of 2,2′-azobis (4-methoxy-2,4-dimethyl valeronitrile) (V-70) (308.43 g/mol) (molar ratio of CPDT: V-70 = 5:1), and benzene (100 mL) into air free round bottom flask.
- Degas the reaction mixture using argon and stir for 30 min. Subsequently put the solution in a temperature-controlled oil bath at 30 °C and react for 18 h.
NOTE: The targeted molecular weight for the Macro-CTA is 10,000 g/mol. 18 hours was determined to be the time necessary to reach reasonable conversion. The color of the polymer solution is transparent light yellow. - After 18 h, terminate the reaction by submerging the round bottom flask in liquid N2.
- Precipitate the polymer by pouring the light-yellow solution of polymer/benzene (~100 mL) into 400 mL of hexane.
- Stir the mixture for 5 min. Precipitate will be settled at the bottom of the beaker and is recovered by filtration.
- Dry the precipitate overnight under vacuum. Then dilute it in 400 mL of tetrahydrofuran (THF). Re-precipitate in hexane.
- Dry this new precipitate again with argon overnight.
NOTE: Macro-CTA is a fine yellow powder. The product yield of the reaction will be ~43.8%. The Mn of the PGMA Macro-CTA is 7,990 g/mol with a polydispersity (PDI) of 1.506 (MW = 12,030 g/mol).
- Synthesis of PGMA-b-PVDMA
- Fractionally distill the VDMA under reduced pressure, and reserve the middle fraction (~70%) for use.
NOTE: This is required to remove polymerization inhibitor. The distillation apparatus is attached to a Schlenk line and the air seal valve is partially opened to the vacuum line. Minimal heat is applied using a varistat and heating mantle until the VDMA monomer begins distilling over at a rate of 1 drop per second. - Combine the 2-Vinyl-4,4- dimethyl azlactone (VDMA) (139.15 g/mol)monomer (10.436 g) with the PGMA-macroCTA (1.669 g), V-70 (14.5 mg; molar ratio of PGMA-macroCTA: V-70 = 3:1) and benzene (75.0 mL) in a single-neck 250-mL round-bottom reaction flask equipped with a Teflon-coated magnetic stir bar.
NOTE: Molecular weight information, PVDMA: 139.15 g/mol, PGMA-macroCTA: 12,030 g/mol, Benzene: 78.11 g/mol. - Degas the mixture with high purity argon and stir for 30 min, and then put in oil bath at 32 °C for 18 h.
- Terminate the reaction by submerging the round bottom flask in liquid N2.
- Precipitate the polymer three times into hexane and dry it at room temperature under vacuum.
- Characterize the molecular weight and PDI of the product by using size exclusion chromatography (S) (see the Table of Materials) according to the procedure in Lokitz et al.20.The size exclusion chromatograph (S) is equipped with three PLgel 5 µm mixed-C columns (300 x 7.5 mm) in series, a refractive index detector (Wavelength= 880 nm), a photodiode array detector, multi-angle light scattering (MALS) detector (Wavelength= 660 nm), and a viscometer (see the Table of Materials).
NOTE: All experiments performed in this manuscript used product with PGMA and PVDMA block lengths of 56 and 175, respectively. The molecular weight of the block copolymer was 37,620 g/mol and the PDI was 1.16.
- Fractionally distill the VDMA under reduced pressure, and reserve the middle fraction (~70%) for use.
2. Generation of Parylene Stencil Patterns Over Silicon Substrates
- Parylene coating
- Sonicate silicon wafers in 50% wt. acetone in water for 5 min followed by sonication in 50% wt. isopropanol (IPA) in water for 5 min.
- Rinse silicon wafers with deionized (DI) water and blow dry with nitrogen gas.
- Deposit 80 nm and 1 µm thick parylene N on 4-inch silicon wafers using a parylene coater (see the Table of Materials).
NOTE: Characterize the thickness of parylene films by using a surface profilometer (see the Table of Materials).- Calibrate parylene film thickness with parylene dimer mass for each individual parylene coating system.
NOTE: In the current system, ~80 mg and ~1000 mg parylene N dimer was required to obtain 80 nm and 1 µm film thickness, respectively (based on the calibration curve obtained). - Use the following settings during operation of the parylene coater: pressure: 80 mTorr, duration: 1 h, furnace temperature: 690 °C, vaporizer temperature: 160 °C.
- Calibrate parylene film thickness with parylene dimer mass for each individual parylene coating system.
- Photolithography
- Bake wafers in an oven at 100 °C for 20 min; then let wafers sit for another 3 min at room temperature.
NOTE: Additional wait time improves adhesion of the photoresist. - Add 2 mL of positive photoresist (see the Table of Materials) and dispense at the center of the parylene-coated wafer. Spin coat the wafers at 3000 rpm for 30 s.
NOTE: Spin coating must be done under the hood. - Wait 1 min, bake wafer on a hot plate at 105 °C for 1 min.
- Load photomask in a mask alignment system (see the Table of Materials). Expose wafers to UV light (λ=325 nm) for 10 s with a dosage of 65 mJ/cm2.
- Let the wafers sit for another 5 min at room temperature.
- Develop wafers by submerging in developer (see the Table of Materials) solution for 2 min. Rinse the wafers with deionized water, and then dry with N2. Do this under the hood.
NOTE: After developing, photoresist appears completely removed from areas exposed to UV. Use an optical microscope (see the Table of Materials) to verify the wafers.
- Bake wafers in an oven at 100 °C for 20 min; then let wafers sit for another 3 min at room temperature.
- Reactive ion etching
- Use a reactive ion etching (RIE) tool (see the Table of Materials) to etch developed wafers with oxygen plasma.
- Apply an oxygen flow rate of 50 cm3/min at a chamber pressure of 20 mTorr.
- For a parylene film thickness of 1 µm, use RF power of 50 W and inductively coupled plasma (ICP) power of 500 W for 100 s was to remove exposed parylene from patterned areas. This corresponded to a parylene etch rate of 1.0-1.15 µm/min.
- For a parylene thickness of 80 nm, use RF power of 50 W and ICP power of 200 W for 55 s to remove exposed parylene from patterned areas. This corresponds to a parylene etch rate of 570-620 nm/min.
NOTE: For efficient parylene removal, determine the parylene etch rate for each RIE system. - Inspect etched substrates with an optical microscope. The silicon surface will appear shiny after the parylene is completely removed from exposed regions.
- Verify etch depth using a surface profilometer (see the Table of Materials).
3. Parylene Lift-off Procedure
- Preparation of polymer solutions
- Dissolve PGMA-b-PVDMA into chloroform (1% wt.). Chloroform should be anhydrous to prevent hydrolysis of azlactone groups.
NOTE: Chloroform is the preferred solvent because it has a high degree of solubility for the polymer, allowing for more uniform surface deposition of single polymer chains compared to other organic solvents25.
- Dissolve PGMA-b-PVDMA into chloroform (1% wt.). Chloroform should be anhydrous to prevent hydrolysis of azlactone groups.
- Cleaning parylene stencils with the plasma cleaner
- Turn on the plasma cleaner (see the Table of Materials) main power and put the parylene-coated substrates in the plasma cleaner chamber.
- Turn on the vacuum pump and evacuate the air in the chamber until the pressure gauge is less than 400 mTorr.
- Slightly open the metering valve and allow the air to enter to the plasma cleaner until the pressure gauge shows 800-1000 mTorr.
- Select RF with Hi mode and expose the substrates for 3 min.
- At the end of process, turn off the RF power and vacuum pump.
- Turn off the plasma cleaner and remove the substrates.
NOTE: After plasma cleaning, the surface shows hydrophilic behavior (Figure 1B) . The water contact angle of bare silicon surfaces before and after plasma cleaning are 27° ± 2° and 0°, respectively.
- Spin-coating of PGMA-b-PVDMA, annealing and sonication over the parylene stencils
- Immediately spin-coat the substrates with 100 µL of 1% wt. PGMA-b-PVDMA in anhydrous chloroform at 1500 rpm, for 15 s using a spin coater (see the Table of Materials).
NOTE: Perform spin-coating within 1-2 s of pipetting the polymer solution to minimize film non-uniformity caused by rapid chloroform evaporation. - Anneal the polymer films at 110 °C in a vacuum oven (see the Table of Materials) for 18 h.
NOTE: Annealing allows for polymer microphase segregation and surface attachment of the GMA block to the surface26.- After the annealing, characterize the polymer coating by measuring the contact angle of substrates. Surfaces show a contact angle of 75°± 1° (Figure 1C)20.
- Sonicate the substrates in 20 mL of acetone or chloroform for 10 min to remove the parylene layer and any physisorbed polymer.
NOTE: Use the following sonication conditions: ultra sonic power, 284 W; Operating frequency, 40 kHz (see the Table of Materials).
NOTE: Parylene can also be peeled off the substrate by applying a piece of Scotch tape at the edge of the substrate then pulling the tape away27. - Store the substrates under vacuum in a desiccator until characterization.
- Immediately spin-coat the substrates with 100 µL of 1% wt. PGMA-b-PVDMA in anhydrous chloroform at 1500 rpm, for 15 s using a spin coater (see the Table of Materials).

Figure 1: Contact angle measurements for treated silicon substrates. (A) Bare silicon, (B) Plasma-cleaned silicon, (C) Spin-coated silicon with PGMA-b-PVDMA (after annealing and sonication in chloroform). Please click here to view a larger version of this figure.
4. PGMA-b-PVDMA Interface-Directed Assembly Procedure
NOTE: This procedure can be performed on substrates containing either a chemically inert background (section 4.1), or a biologically inert background (section 4.2), depending on the application.
- Preparation of chemically inert background on silicon substrates
- Use oxygen plasma cleaner to clean the bare silicon (section 3.2).
- Pipette 100 µL of trichloro(1H,1H,2H,2H-perfluorooctyl) silane (TPS) onto a Petri dish and place the silicon substrates inside a vacuum desiccator next to the Petri dish.
- Apply vacuum (-750 Torr) for 1 h for chemical vapor deposition (CVD).
CAUTION: TPS is highly toxic and the CVD process should be performed inside a fume hood.
NOTE: After 1 h the substrate shows hydrophobic behavior. A contact angle of 109°± 3° is typically measured after the CVD process. The thickness of the TPS film is 1.5 ± 0.5 nm.
NOTE: TPS blocks reaction of the reactive surface oxide with PGMA-b-PVDMA. - Coat the wafers with parylene (1 µm thickness). Perform photolithography and reactive ion etching to generate parylene patterns (section 2) and to etch away the TPS layer in the exposed regions.
- Preparation of polyethylene glycol (PEG) background on silicon substrates.
- Use the oxygen plasma cleaner for 3 min to clean the bare silicon substrates (section 3.2).
- Perform CVD of TPS for 1 h (section 4.1.2).
- Immerse substrates into a 0.7% wt/v solution of Pluronic F-127 in ultrapure water for 18 h to generate a PEG layer on the surface28,29.
NOTE: Pluronic contains a hydrophobic polypropylene oxide (PPO) polymer block between two PEG chains. The PPO block anchors the polymer to the TPS surface while the PEG chains are exposed to solution28. - Wash and rinse the substrate for 5 min with 100 mL of ultrapure water.
- Deposit 80 nm and 1 µm thick parylene N on 4-inch silicon wafers using a parylene coater.
- Perform photolithography and reactive ion etching to generate parylene patterns (section 2).
- Sonication, spin-coating of PGMA-b-PVDMA polymer, and annealing the substrates
- Sonicate chemically inert (TPS) substrates (section 4.1) or PEG-functional substrates (section 4.2) for 10 min in acetone to remove the parylene layer.
- Spin-coat the sonicated substrate with 100 µL of 1% wt. PGMA-b-PVDMA in anhydrous chloroform at 1500 rpm for 15 s.
- Anneal the polymer films at 110 °C under vacuum for 18 h.
- Sonicate the substrates in acetone or chloroform for 10 min to remove physisorbed polymer present in background regions on the surface.
- Store the substrates in a vacuum desiccator until further use.
5. Custom PGMA-b-PVDMA Micro-Contact Printing (μCP)
- PDMS stamp fabrication
- Fabricate the silicon masters according to the standard photolithography procedure30. Use CVD process (section 4.1.2) to deposit anti-adhesive TPS onto the silicon masters.
NOTE: The silicon mold should be treated with TPS the first time it is used, and re-applied after it has been used 5-10 times. - Perform standard soft lithography methods for fabrication of stamps (PDMS precursor to curing agent mass ratio 10:1)31.
NOTE: Stamps used in this study consist of micropillar arrays (diameter = 5-50 µm, height = 20 µm). - Cut out a single stamp. Clean the stamp by sonicating for 10 min in HCl (1 M), 5 min in acetone, followed by 5 min in ethanol.
- Dry the stamps in a convection oven at 80 °C for 20 min to remove residual organic solvent.
- Fabricate the silicon masters according to the standard photolithography procedure30. Use CVD process (section 4.1.2) to deposit anti-adhesive TPS onto the silicon masters.
- Microcontact printing of PGMA-b-PVDMA onto silicon substrates
- Deposit TPS onto the surface of PDMS stamps using the CVD process (section 4.1.2).
NOTE: The TPS layer is used to prevent coupling of the polymer to the stamp surface.
NOTE: Contact angle measurements can be used to characterize stamps after TPS adsorption, as shown in Figure 2 (Inset A, B). - Dissolve the PGMA-b-PVDMA polymer into anhydrous chloroform at a concentration of 0.25-1% wt.
- Submerge the stamps into 5 mL of the polymer solution for 3 min.
- Plasma clean 2×2 cm bare silicon substrates for 3 min to clean surface for coupling with the PGMA blocks (section 3.2).
- Take out the polymer-coated stamps from the polymer solution.
NOTE: Stamps must be used for printing while they are still wet and a layer of solution exists over them. - Put inked stamp directly on silicon substrate.
- Use a manual drill press stand (see the Table of Materials) (Figure 3) to press the polymer-coated stamps onto the silicon surface to promote pattern transfer. Immediately apply the stamp to the substrate (within 1-2 s) after taking out the coated stamps from polymer solution.
NOTE: Both the silicon and the PDMS stamp can be placed on double-sided tape backing to minimize PDMS stamp deformation due to non-uniform or high pressure stamping32. - Apply conformal contact between polymer-inked stamp and silicon substrate for 1 min. Use the estimated pressure of 75 g/cm2(7.35 kPa) to press.
- Gently separate the stamp from the silicon surface.
- Anneal the printed silicon substrates immediately in a vacuum oven at 110 °C for 18 h.
- Sonicate the printed silicon substrates in acetone or chloroform for 10 min to remove any physically-adsorbed PGMA-b-PVDMA and then dry with N2.
- Perform surface characterization analysis for both PDMS stamp (after printing step) and printed-silicon (after annealing and sonication steps) to verify the successful transfer of PGMA-b-PVDMA.
NOTE: Surface profilometer and attenuated total reflectance Fourier-transform infrared spectroscopy (ATR-FTIR) analysis could be used to analyze the printed-silicon substrate and PDMS stamp, respectively.
- Perform surface characterization analysis for both PDMS stamp (after printing step) and printed-silicon (after annealing and sonication steps) to verify the successful transfer of PGMA-b-PVDMA.
- Store the substrates under vacuum in a desiccator until characterization.
- Deposit TPS onto the surface of PDMS stamps using the CVD process (section 4.1.2).
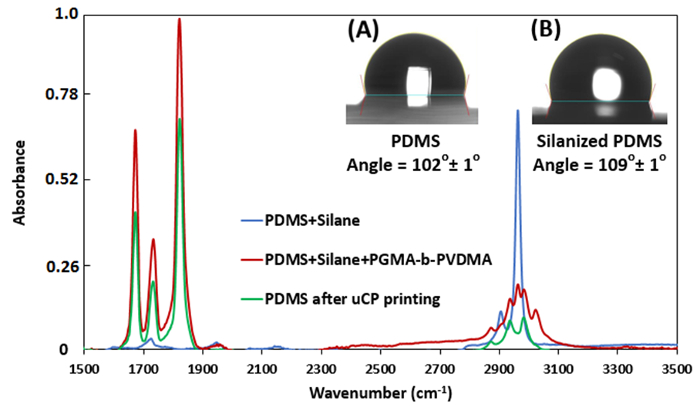
Figure 2: ATR-FTIR measurements for treated PDMS stamps (Relative intensity). (Inset A) Contact angle measurements for bare PDMS stamp. (Inset B) Contact angle measurements for TPS treated PDMS stamp. Please click here to view a larger version of this figure.

Figure 3: Setup for μCP of PGMA-b-PVDMA solutions onto silicon substrates. The procedure includes use of a (A) manual drill press, (B) a TPS-functionalized PDMS stamp coated with the PGMA-b-PVDMA polymer, (C) a plasma cleaned 2×2 cm silicon substrate, and (D) double-sided tape.
Contact angle measurements can be used to evaluate the functionalization of silicon with PGMA-b-PVDMA. Figure 1 depicts the contact angle of the silicon substrate during the different processing steps. Hydrophilic behavior of the plasma cleaned silicon substrate is shown in Figure 1B. The contact angle after polymer spin coating and annealing is 75° ± 1°(Figure 1C) which is consistent with the values reported by Lokitz et al. for PVDMA surfaces20.
Figure 2 shows the ATR-FTIR spectra and contact angle measurement of PDMS stamps during the different steps of the µCP procedure. After printing, the azlactone carbonyl stretching vibration at ~1818 cm-1 decreases by 34 9%. Figure 2 (inset A, B) also depicts the change in hydrophobicity of PDMS stamps after TPS treatment.
Stamp-substrate pressing is a critical step in µCP. Figure 3 exhibits different parts of the manual rotary tool necessary to achieve uniform contact between the polymer-coated stamp and silicon substrate.
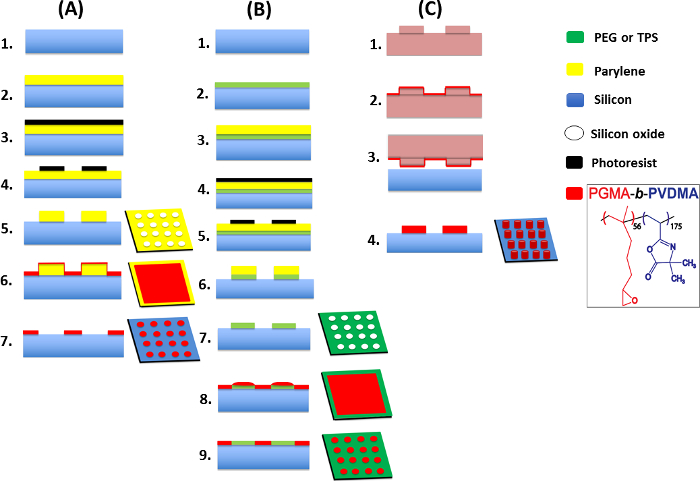
Figure 4: Details of the developed techniques for generating PGMA-b-PVDMA into patterned, crosslinked or brush films. This figure has been modified from Masigol et al.24. (A) Schematic representation of the parylene lift-off protocol for patterning polymer brushes onto silicon substrates, 1. silicon wafer (w/native oxide), 2. parylene deposition (1 µm or 80 nm), 3. photoresist spin coating, 4. UV exposure and development, 5. oxygen plasma etching, 6. polymer spin coating, 7. annealing and parylene lift-off. (B) IDA procedure for patterning polymer brushes onto biological/chemical (PEG/TPS) inert substrates, 1. silicon wafer (w/native oxide), 2. PEG/TPS deposition, 3. parylene deposition (1 µm or 80 nm), 4. photoresist spin coating, 5. UV exposure and development, 6. oxygen plasma treatment, 7. parylene lift-off, 8. polymer spin coating, 9. annealing and sonication. (C) Generation of crosslinked polymer structures onto silicon using the µCP method, 1. soft-lithography for making PDMS stamp followed by TPS coating, 2. polymer inking on TPS-functionalized PDMS, 3. stamp/substrate contact, 4. annealing and sonication. Please click here to view a larger version of this figure.
Figure 4 shows the step-by-step procedures for generating polymer patterns24. These procedures are designed to: (1) pattern uniform brush structures of PGMA-b-PVDMA polymers onto chemically/biologically inert substrates by applying parylene lift-off and IDA techniques (Figure 4A, 4B), or (2) generate thicker film patterns of micron-scale thickness (Figure 4C).
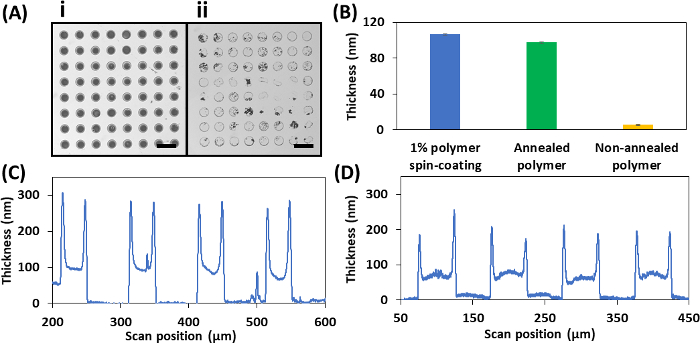
Figure 5: Representative results of the parylene lift-off procedure. (A) Brightfield images of PGMA-b-PVDMA polymer patterns on silicon with annealing (inset i) and without annealing (inset ii) (Scale bar = 40 µm). (B) Polymer thickness measured after 10 min sonication in chloroform with or without annealing. (C) Cross-sectional polymer height profile for 1 µm thick parylene stencils. (D) Cross-sectional polymer height profile for 80 nm thick parylene stencils. Please click here to view a larger version of this figure.
The parylene lift-off technique can be used to achieve brush structures of PGMA-b-PVDMA block co-polymers, corresponding to ~90 nm film thickness. Figure 5A (inset i) depicts the patterned spots surrounded by polymer-free background. Annealing is the crucial step leading polymer phase-segregation and strong covalent surface attachment through reaction of epoxy groups on the GMA block with surface oxide24. As Figure 5A (inset ii) shows, without annealing, sonication in chloroform will remove much of the patterned polymer. To investigate the effect of annealing in more detail, a 1% wt. concentration of polymer in chloroform was spin-coated over a plasma-cleaned silicon substrate (without parylene). Polymer thickness was measured by ellipsometry (see the Table of Materials). While sonication in chloroform led to the removal of most of the polymer from non-annealed substrates, no significant change in thickness of polymer was observed for annealed substrates (Figure 5B). Compared to 1 µm parylene stencils, 80 nm parylene stencils generated higher film uniformity (Figure 5C, 5D).
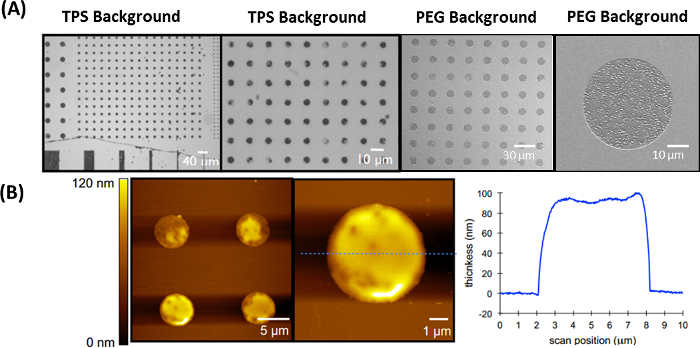
Figure 6: Representative results of the IDA method for generating brush-like patterns of PGMA-b-PVDMA in chemically and biologically inert backgrounds. This figure has been modified from Masigol et al.24. (A) PGMA-b-PVDMA patterns in TPS and PEG backgrounds. (B) AFM measurement of polymer patterns and representative polymer film thickness over TPS-coated substrates. Please click here to view a larger version of this figure.
The IDA technique can be used to co-pattern uniform films of the PGMA-b-PVDMA polymer over chemically or biologically inert backgrounds. Figure 6A shows the PGMA-b-PVDMA patterns on PEG/TPS backgrounds. This approach results in patterned films of 90-100 nm thickness without the edge defects observed from the prior method (Figure 5C, 5D). AFM profiles in Figure 6B depict polymer film thicknesses obtained using the IDA method.
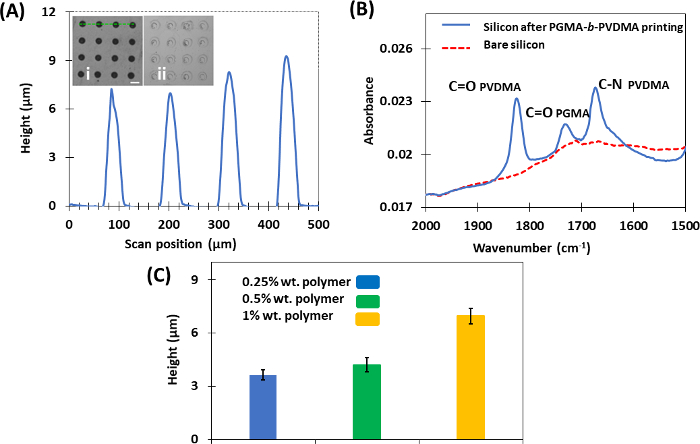
Figure 7: Representative results of μCP technique for making cross-linked films of PGMA-b-PVDMA. This figure has been modified from Masigol et al.24. (A) Height profiles of polymers printed on the silicon substrates (1% wt. polymer). (inset i) PGMA-b-PVDMA patterns obtained after µCP with annealing, and (inset ii) without annealing (scale bar = 30 µm). (B) ATR-FTIR analysis of bare silicon and silicon substrate after PGMA-b-PVDMA printing. (C) Effect of using different polymer inking concentrations on the average crosslinked film height (Error bars describe standard deviation from the average). Please click here to view a larger version of this figure.
µCP was developed as the final approach to patterning PGMA-b-PVDMA polymers on silicon surfaces. In contrast to parylene lift-off and IDA techniques, this approach results in polymer films patterned at micron-scale thickness (Figure 7A). There were several critical steps that were required to insure efficient transfer of polymer from the stamp to the substrate during the printing process. First, PDMS functionalization with TPS was required to inhibit PGMA-b-PVDMA coupling to the stamp (Figure 2, inset A, B). Second, plasma treatment on the substrate was required to form an oxide surface layer for reaction with epoxy groups present in the PGMA block of the polymer (Figure 1B). Finally, annealing of the stamped polymer films was required to promote crosslinking throughout the film; Figure 7A (inset i and ii) show annealed and non-annealed substrates after sonication, where significant damage to the non-annealed films was observed. Another requirement for the patterning technique was to preserve the azlactone functionality, which was verified by measuring the carbonyl stretching vibration at ~1818 cm-1 (Figure 7B). Finally, the µCP technique also enabled microscale control of polymer thickness films by varying the concentrations of PGMA-b-PVDMA in chloroform during the inking step (Figure 7C).
