1. Ex vivo infection
- Medium and equipment
- Under sterile conditions, prepare fresh Drosophila Hemocyte Isolating Medium (DHIM) containing 75% Schneider's Drosophila medium with 25% Fetal Bovine Serum (FBS) and filter sterilize it.
- Layer 2-3 pieces of 10 cm x 10 cm paraffin film under a stereomicroscope.
- Prepare the glass capillary. Set the capillary puller heater to 55% of maximum. Pull the capillary tube to a sharp point of approximately 10 µm.
- Backfill the capillary with mineral oil.
- Assemble filled capillary tube onto the nanoinjector (Figure 1A), and open fused capillary tube tip by breaking off the tip with forceps (Figure 1B). The outer diameter of the tip should be 100 µm for easy uptake of the hemocytes.
- Eject as much oil as possible from the capillary tube tip, then fill with DHIM. For easy visualization of the border of up-taken hemolymph and the oil, include an air bubble between the oil and DHIM (Figure 1B').
- Hemolymph extraction
- Pick 3rd instar Drosophila larvae from the inside wall of food vials gently using forceps and place them into a 100 µm strainer (Figure 1C). 3rd instar larvae are found 3-6 days following fertile egg laying by an adult female.
NOTE: These experiments used the genotype, w1118;P{w+mC=Hml-GAL4.Δ}2,P{w+mC=UAS-2xEGFP}AH2, since hemocytes from these animals express enhanced green fluorescent protein (EGFP) to aid in identification of the cells by microscopy. - Pour 5 mL of sterile water over larvae and shake the strainer for 5 s. Place the strainer onto task wipe to remove the excess water (Figure 1C').
- Transfer the larvae into a 1.5 mL microcentrifuge tube. Anesthetize them with CO2 gas for 5 s (Figure 1D).
- Place the larvae onto paraffin film under the stereomicroscope, with dorsal-side facing up (Figure 2A).
- Place the glass capillary lightly onto the larval posterior body to hold it in place and disrupt the posterior cuticle open using fine pointed forceps (Figure 2B).
- Allow the hemolymph to flow onto the paraffin film (Figure 2C).
- Make a pool of hemolymph including hemocytes from 20-50 larvae at a time on the paraffin film.
- Take up pooled hemolymph using the glass capillary on the nanoinjector (Figure 2D).
NOTE: There should be approximately 10-20 µL of hemolymph. - Eject the hemolymph into a 1.5 mL microcentrifuge tube containing 500 µL of DHIM (Figure 2E).
- Repeat steps 1.2.4) – 1.2.9) for every batch of larvae.
- Pick 3rd instar Drosophila larvae from the inside wall of food vials gently using forceps and place them into a 100 µm strainer (Figure 1C). 3rd instar larvae are found 3-6 days following fertile egg laying by an adult female.
- Count the number of hemocytes.
- Pipette 5 µL of 0.4 % Trypan blue solution in a 0.6 mL microcentrifuge tube to stain the dead cells. Gently mix the DHIM and hemocytes in the 1.5 mL tube using a pipette, and transfer 5 µL of DHIM including hemocytes to the 0.6 mL microcentrifuge tube and mix gently.
- Pipette 10 µL of cells from the 1:1 Trypan blue:hemocyte mixture into the hemocytometer.
- Count the number of live hemocytes that are not stained with Trypan blue in each of the 4 corner fields of the hemocytomter and calculate the concentration of the hemocytes per milliliter using formula:
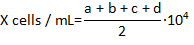
where X is the concentration of live hemocytes per milliliter; a, b, c, and d are the number of live cells (as determined by Trypan blue exclusion) in each 4 of the fields counted in the hemocytometer. Division by 2 of the total number of cells counted is due to the 1:1 dilution of the cells with Trypan blue. Cells stained with Trypan blue are considered dead.
- Ex vivo Infections
- Determine the number of wells to be seeded with cells based on timepoints and biological replicates needed for each experiment. 5.0×104 hemocytes are desirable for RNA and protein purification following infection.
- Calculate the volume of pathogen stock to be diluted with DHIM for infection using the following formula:
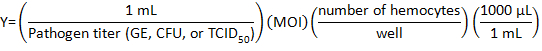
where Multiplicity of infection (MOI) is the number of viral or bacteria desired per cell.
NOTE: MOI used depends on the individual experiment and assays performed. Here, 10 genome equivalents (GE)/cell of C. burnetii, 10 CFU/cell of Listeria, or 1 TCID50/cell of IIV6 was used. - Prepare 500 µL of pathogen medium for each well of a 24-well plate by adding the proper volume of DHIM to the viral or bacterial volume in a tube.
- Place a 12 mm round cover glass (no. 1 thickness) in a well of a 24-well plate.
- Split the DHIM including the hemocytes into wells of a 24-well plate.
- Add 500 µL of pathogen medium to hemocytes in a well.
- Centrifuge the plate at 1,000 x g for 5 min.
- Incubate the plate for 1 h at 28 °C. Every 15 min, gently tilt the plate from back to front, then left to right for 5 s by hand.
- After the 1 h of invasion/attachment step 1.4.8), gently pipette off the pathogen medium and wash the hemocytes with fresh DHIM, and refill it with 500 µL of fresh DHIM.
- Incubate the infected hemocytes for the desired time. In these experiments, C. burnetii-or IIV6-infected hemocytes are incubated for 24 h, and Listeria-infected hemocytes are incubated for 1, 2, or 4 h.
2. In vivo infection
- Infection
- Warm the Drosophila fruit juice agar plate at room temperature for 15 min. Plates are made as previously described20.
- Add 30 g of agar to 700 mL of water and autoclave it for 40 min.
- Dissolve 0.5 g of methyl paraben in 10 mL of absolute ethanol.
- Add the methyl paraben solution to 300 mL of fruit juice concentrate.
- Quickly mix the juice concentrate into the autoclaved agar solution and dispense 5 mL into 10 × 35 mm Petri dishes.
- After the plates have cooled for 15 min, store them at 4 °C.
- Prepare 3rd instar larvae following steps 1.2.1) – 1.2.3).
- Place the yeast paste on an agar plate. Make a fine cut in the agar plate where larvae can migrate to avoid drying (Figure 3A). Assemble a 0.001 mm pointed tungsten needle with holding forceps using paraffin film (Figure 3B, C).
- Pipette 50 µL of high-titer mCherry expressing-C. burnetii (5.95×109 GE/mL) onto the paraffin film under the stereo microscope and place the larvae into the pool of bacteria.
- Place the larvae into the pool of bacteria. Prick the larvae with a tungsten needle (Figure 3D). Transfer the larvae onto an agar plate (Figure 3E).
- Transfer the remaining pathogen medium onto the agar plate and seal the plate with paraffin film.
- Keep the larvae on the plate in moist air until the desired time post-infection (Figure 3F). In this experiment, C. burnetii-infected larvae are on the plate for 24 h.
- Warm the Drosophila fruit juice agar plate at room temperature for 15 min. Plates are made as previously described20.
- Hemolymph extraction and plating of hemocytes
- Prepare the medium and equipment following step 1.1).
- Place a 12 mm round cover glass (no. 1 thickness) in a well of a 24-well plate. Pipette 500 µL of DHIM into the well.
- Extract the hemolymph from the infected larvae following steps 1.2.4) – 1.2.8).
- Eject the hemolymph into the well following step 2.2.2).
- Repeat 2.2.3) and 2.2.4) for multiple batches of larvae.
- Centrifuge the plate at 1,000 x g for 5 min.
3. Visualization
- Fixing and staining
- After allowing the hemocytes to settle on the round cover glass in the well, gently remove the medium from each well.
- Gently add 200 µL of 4% paraformaldehyde (PFA) to each well of settled hemocytes. Incubate the hemocytes for 20 min at room temperature.
- Remove the 4% PFA and gently add 200 µL of PBS containing 0.1% Triton X-100 and 1% Bovine Serum Albumin (BSA) to each well. Incubate the hemocytes for 10 min at room temperature.
- Remove the PBS and gently add 200 µL of 1× 4',6-diamidino-2-phenylindole (DAPI) to each well. Incubate the hemocytes for 10 min in the dark at room temperature.
- Remove the DAPI solution and gently add PBS to each well. Incubate the hemocytes for 5 min at room temperature.
- Drop 10 µL of the antifade mounting medium on a glass microscope slide.
- After removing the PBS from each well, remove the cover glass from the 24-well plate using fine-pointed forceps. Gently place the cover glass onto the antifade mounting medium on the glass slide, with the hemocytes facing down.
- Allow the slide to dry by placing it in the dark overnight.
- Confocal Imaging
- Configure the confocal microscope for three color imaging of DAPI, EGFP, and mCherry. Use the following setting: DAPI excitation (ex) 405 nm, emission (em) 415-480 nm; EGFP ex 488 nm, em 493-564 nm; mCherry ex 587 nm, em 597-700 nm.
- Place the sample on the microscope and focus on the sample using a 63X/1.4 numerical aperture (NA) objective. Locate desired hemocytes in the field of view for imaging.
- Adjust laser power and detector gains to achieve appropriate exposure of the sample. Check multiple z-planes to ensure the exposure level is appropriate for the entire sample thickness.
- Find the top and bottom position on the z-axis of a whole hemocyte. Set these positions as the start and end positions for z-sectioning.
- Only use scanning zoom to image the area containing the hemocyte. Zoom factors of 3X are often used.
- Collect the image series at appropriate resolution such as 1024 x 1024 pixels in the x-y plane, and 0.3 µm spacing in the z dimension.
- 3D model reconstruction
NOTE: Open source software exists that performs many of the functions described below for 3D model reconstruction.- Import the z-sectioned image series file into the software associated with the confocal microscope for 3D model reconstruction.
- Select a cell showing co-localization of nuclei stained with DAPI and mCherry expressing C. burnetii in an EGFP expressing hemocyte. Crop the image series to contain only the single cell.
- Select the 3D Viewer option which reconstructs the 3D model using the software's pre-packaged algorithm. Choose the desired type of 3D representation among Blend, Surface, and Mixed options. In this method, hemocytes, nuclei, and C. burnetii are shown using surface models.
- Observe the 3D reconstructed cell from various viewing positions by holding the mouse button and dragging the cursor around the screen. Adjust the cell orientation and position of the modeled light source to optimize the image. Other options for Opacity, Minimum and Maximum Threshold, Specular, Ambient, Shineness, and Gamma exist to optimize the image.
- Take cross-sections through the model using clipping and sectioning commands to visualize the interior contents of the hemocyte.
4. Application for gene and/or protein analysis
- Following the infection with IIV6 and Listeria, lyse cells for subsequent qRT-PCR or Western blot analysis as previously described21 and following manufacturer's instructions.
NOTE: Refer the Table of Materials for the primers for qRT-PCR and the antibodies for Western blot. - Analyze PCR products by agarose gel electrophoresis, as previously described22, to ensure the proper length of the amplified product.
To collect live hemocytes for ex vivo infection, up to 3×106 hemocytes were extracted from 200 Drosophila 3rd instar larvae. To develop our method, a number of different techniques were attempted. Individual larval dissection would take up to 1.5 h, and an average of ~8000 cells were obtained using this method18, most of which were not alive by the end of collection. Next, we tried to extract hemolymph, which contained the hemocytes, from 20 larvae at a time using a glass capillary tube19, but the capillary became clogged with cuticle material and the hemolymph was not able to be efficiently taken up by the glass capillary. Finally, we mechanically disrupted the cuticle of 20-50 larvae per batch with fine forceps12, and made a pool of hemolymph for easy uptake. This allowed easy collection of a large amount of hemocytes from a large number of larvae (Table 1).
To indicate that the extracted hemocytes were alive and suitable for the infection, a Trypan blue exclusion assay was performed to calculate the percentage of live hemocytes extracted using the method presented here (Figure 6A). In addition, we compared our method for hemocyte extraction with a previously published method where the goal was to develop a mechanical disruption method to isolate and differentiate between circulating and resident hemocytes from individual Drosophila larva23. Upon comparison between the two methods from 10 independent experimental isolations, we observed that cell viability was slightly greater using the method presented here (Figure 6A); however, the method from Petraki et al. yielded close to twice as many hemocytes from every 10 dissected larvae (Figure 6B). A reason for the increased number of hemocytes from the Petraki et al. method is that this method collects resident (sessile) hemocytes, as well as circulating hemocytes. To confirm that the cells extracted with the method presented here were in fact hemocytes, we utilized a fly line containing transgenes for the hemolectin (Hml) promoter driving GAL4 in circulating hemocytes that binds the upstream activator sequence (UAS) to activate enhanced green fluorescent protein (EGFP) transcription and expression in the hemocytes. Hemocytes from hml-GAL4>UAS-EGFP larvae were extracted onto chambered coverglass slides, fixed, permeabilized and stained with DAPI as previously described21. Figure 7 shows that most DAPI-positive cells are also EGFP-positive. Quantification of co-expression was performed from multiple images, from which we calculated that 86±9% of cells isolated were hemocytes.
The protocol in innovative 3D models of pathogen invasion into Drosophila hemocytes extracted from 3rd instar larvae following ex vivo or in vivo C. burnetii infection. We were able to image C. burnetii infection since the bacteria express mCherry. After infected hemocytes were fixed and stained, they were imaged for DAPI, EGFP, and mCherry using confocal microscopy. Hemocyte infection rates, as determined by the percentage of EGFP-positive cells that also exhibited mCherry signal, for both ex vivo and in vivo C. burnetii infections was nearly 100%. This was expected since an MOI of 10 GE/cell was used for ex vivo infection, which should result in infection of 100% of cells24. Regarding the in vivo infections, since larvae are placed in a high-titer droplet of mCherry expressing-C. burnetii (5.95×109 GE/mL), the number of bacteria is roughly 10,000-fold higher than the number of hemocytes per larva. Therefore, a 100% infection rate is expected for the in vivo infections.
Next, Z-sections were collected through the hemocyte to visualize the entire cell in 3D, and to confirm the presence of C. burnetii in the interior of the cell. Figure 4 shows transparent cross-sections of a hemocyte (in green) with C. burnetii (in red) seen in the interior of the cell. The images were also reconstructed into a 3D model (Figure 5) representing the surfaces of the hemocyte and C. burnetii, again showing C. burnetii in the interior of the cross-sectioned hemocytes. Interestingly, in vivo-infected hemocytes exhibited greater cytoplasmic extensions and were flatter in nature, while ex vivo-infected hemocytes were more spherical (Figure 5). This could indicate a greater population of lamellocytes in the in vivo-infected population and plasmatocytes in the ex vivo population7. While lamellocyte differentiation is generally induced during parasitic wasp infection25, wounding of the Drosophila larvae is also sufficient to induce lamellocyte differentiation26. Finally, while not utilized in the experiments presented here, there are GFP-expressing forms of Listeria27 and IIV628 that could be used to generate 3D models of hemocyte infection. Instead, Listeria– and IIV6-infected hemocytes were used for Western blotting and qRT-PCR experiments.
Using the method presented here, infections are performed both ex vivo and in vivo for imaging. In addition, pathogen mRNA and protein analysis followed ex vivo infections. Specifically, extracted hemocytes were infected with IIV6 and were applied to qRT-PCR analysis. It showed significantly higher levels of IIV6-193R, a viral gene that encodes for a putative inhibitor of apoptosis29, between mock- and IIV6-infected cells (Figure 8A). Electrophoresis of the amplified products confirmed the presence of a band for the IIV6-193R gene product in the infected sample, but not the mock-infected sample (Figure 8B). Amplified bands for the RpII endogenous control were found in all samples, and IIV6 infection in S2 cells were performed as a positive control. Since IIV6 infections were performed at an MOI of 1 TCID50/cell, approximately 50% of the cells were expected to be infected, based on the Poisson distribution24.
Total protein from mock- or Listeria-infected hemocytes was collected at 1, 2, and 4 h post-infection and protein concentration was determined by Bicinchoninic acid (BCA) assay. 100% of the cells were expected to be infected ex vivo since the MOI was 10 CFU/cell24. Western blotting confirms the presence of Listeria-derived protein products in the infected hemocytes, with high levels achieved by 4 h post-infection (Figure 9). Listeria inoculum is used as a positive control for the detection of Listeria-specific bands. The presence of actin is shown in the hemocyte samples to confirm levels of protein loading. Taken together, these results indicate that the hemocytes extracted using the method presented here were suitable for both viral and bacterial infection experiments.

Figure 1: Outline of equipment and materials used for hemocyte extraction. Equipment was first prepared prior to dissection. A) The pulled glass capillary was inserted into the nanoinjector after being back-filled with mineral oil. B) The fused tip of the glass capillary was broken with forceps to create a 100 µm outer diameter. B') DHIM was taken up by the capillary to avoid cell contamination and air bubbles were introduced to make a clear distinction between oil and DHIM. C) Larvae are picked from food vials and placed into a cell strainer. C') Larvae are washed in sterile water and water is removed with a task wipe, D) Larvae are transferred into a microcentrifuge tube and anesthetized with CO2 gas. Please click here to view a larger version of this figure.

Figure 2: Timeline and flow chart for the extraction of hemocytes. A) Larvae are placed on their dorsal side prior to opening the cuticle. B) The cuticle is disrupted with fine pointed forceps and the capillary needle. C) The hemolymph is bled onto paraffin film. D) Pools of hemolymph from 20-50 larvae are taken up with the glass capillary and nanoinjector. E) The hemolymph and hemocytes are transferred into a microcentrifuge tube containing 500 µL DHIM to be counted with a hemocytometer. Please click here to view a larger version of this figure.
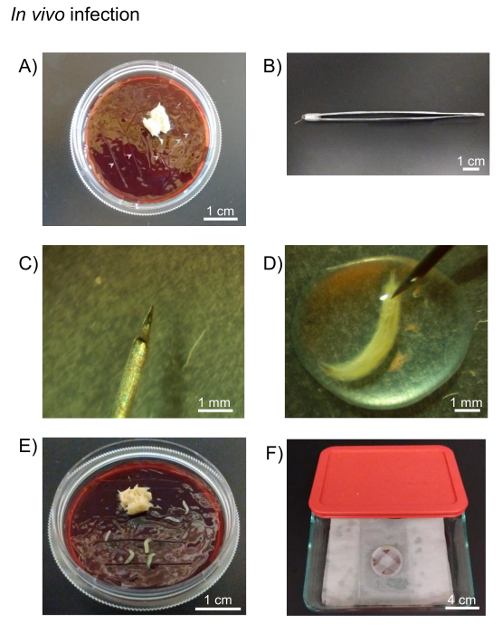
Figure 3: In vivo infection. A) Drosophila fruit juice agar plates and yeast paste are used for incubation of in vivo infected larvae. Cuts are made in the agar plate to facilitate larvae longevity (grey arrows). B) A 0.001 mm pointed tungsten needle is attached to forceps using paraffin film. C) The tip of 0.001 mm pointed tungsten needle. D) The larva is pricked with tungsten needle in the pathogen pool on the paraffin film under stereo microscope. E) The pricked larvae are placed onto the agar plate. F) The plate is sealed with paraffin film and kept on moist paper towels in the container until appropriate time. Please click here to view a larger version of this figure.
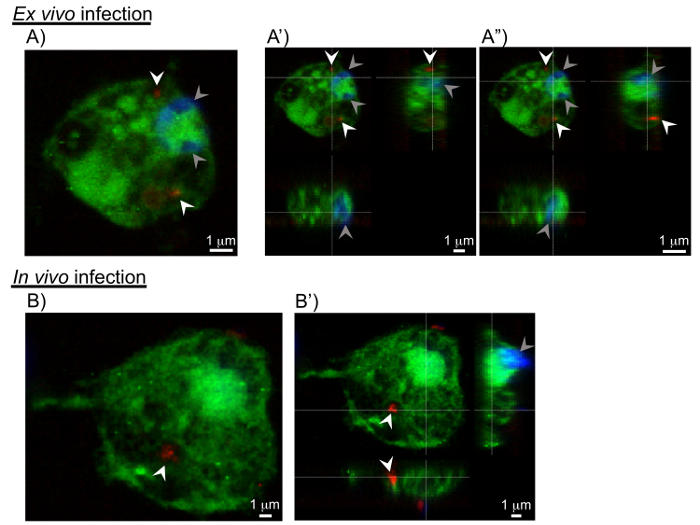
Figure 4: Cross-sections of pathogen invasion into the hemocytes. Sections extracted from 3D confocal scanning of pathogen-infected hemocytes. A, B) The xy-sections shows C. burnetii (in red marked with white arrowheads) in the interior of the hemocyte. The grey arrowheads show the nuclei of hemocytes. A', A", B') Views including yz- and xz-section images show the invasion of C. burnetii into the hemocytes from 2 additional viewing points. Please click here to view a larger version of this figure.
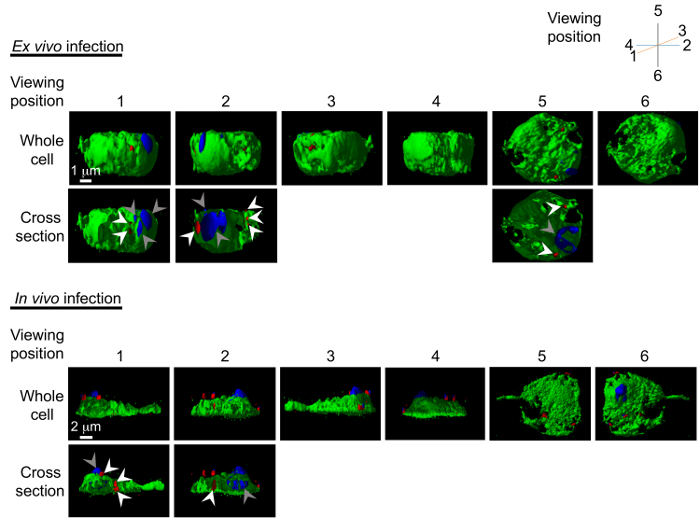
Figure 5: 3D models of pathogen invasion into the hemocytes. Reconstructed 3D models of C. burnetii invasion into the hemocytes are generated following ex vivo and in vivo infection. Model can be freely rotated using the software; here 6 viewing points are shown with the hemocyte in green, C. burnetii in red (white arrowheads), and nucleus in blue (grey arrowheads). The cross-section images showing the interior of pathogen-invaded cells are also shown. Please click here to view a larger version of this figure.
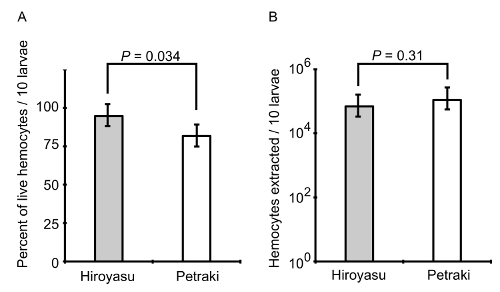
Figure 6: The percentage and population of live hemocytes. Hemocytes were extracted from groups of 10 3rd instar larvae using the method of Petraki et al. and the method presented here. A) The percentage of the live hemocytes was calculated for each technique by Trypan blue exclusion assay. B) The total population of hemocytes was compared between the two techniques. The bar graphs represent the mean ± standard deviation from N = 10 biological replicates per method of extraction and assay type. P-values are indicated from a two-tailed Student's T-test assuming unequal variance. Please click here to view a larger version of this figure.
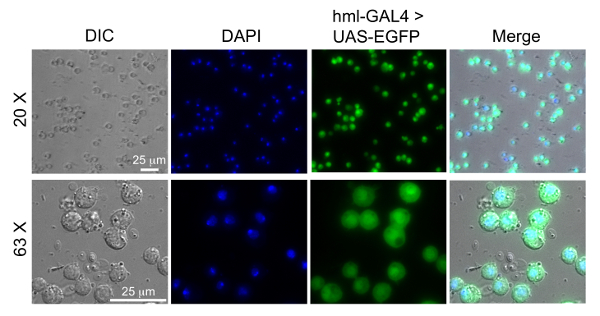
Figure 7: Images of extracted hemocytes using the hemolectin driver. Hemocytes were extracted from 80 larvae of w1118;P{w[+mC]=Hml-GAL4.Δ}2, P{w[+mC]=UAS-2xEGFP}AH2. The promoter for Hml drives GAL4 in circulating hemocytes, which binds UAS to activate EGFP transcription and expression. The hemocytes were fixed and stained with DAPI as previously described20. Hemocytes are mounted on chambered coverslips and imaged by differential interference contract (DIC) and fluorescence microscopy. The blue channel shows the nucleus stained with DAPI and the green channel shows the hemocytes expressing EGFP. Please click here to view a larger version of this figure.
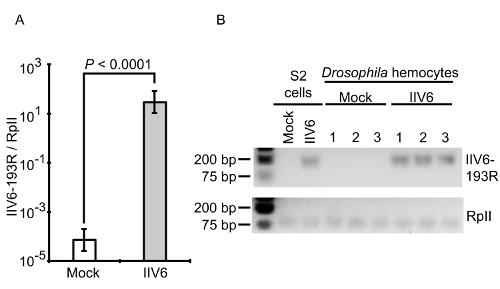
Figure 8: qRT-PCR and gel electrophoresis. The expression of the IIV6-193R gene was observed by qRT-PCR only in the IIV6-infected hemocytes. A) 193R gene expression was normalized to the internal control, RpII, and presented as a ratio. The cycle number for the mock-infected hemocytes was arbitrarily set to a maximum cycle number of 40 for analysis since 193R was not detected in these samples. Data represents the mean ± standard deviation from N = 3 biological replicates per group. The P-value indicates a two-tailed Student's T-test assuming unequal variance. B) The qRT-PCR products are shown by agarose gel electrophoresis. Please click here to view a larger version of this figure.
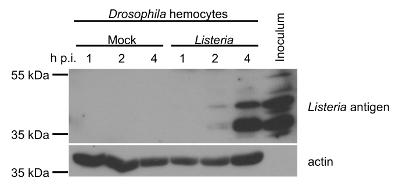
Figure 9: Western Blot Analysis. The expression of Listeria antigens and actin in mock- and Listeria-infected hemocytes from 3rd instar Drosophila larvae at 1, 2, and 4 h post-infection (p.i.) are determined by Western blotting. Total protein concentrations were determined by the Bicinchoninic acid (BCA) assay to normalize total amount of protein loaded in each lane of the gel. Inoculum used to infect the hemocytes was used as a positive control sample. Please click here to view a larger version of this figure.
| Method | Average number of larvae for hemocyte extraction |
Average number of total hemocytes |
| hemolymph capillary extraction (this method) |
192.44 (SD: 86.05) | 4.56 x 105 cells (SD: 5.83 x 105) |
| (Range: 70 – 390, N = 25 trials) | (Range:1.20 x 105 – 2.95 x 106 cells) | |
| individual larvae dissection |
26.67 (SD: 11.55) | 8.33 x 103 cells (SD: 6.66 x 103) |
| (Range: 20 – 40, N = 10 trials) | (Range: 4.00 x 103 – 1.60 x 104 cells) | |
| larval capillary extraction* |
N/A* | N/A* |
| *hemocytes were unable to be extracted using this method due to clogging of the capillary needle | ||
Table 1: Number of dissected larvae and extracted hemocytes using different techniques. The average numbers of dissected larvae and extracted hemocytes were compared between the method presented here and other methods. Our method resulted in collection of higher numbers of larvae and hemocytes. The larval capillary extraction method was also attempted, but hemocytes were unable to be extracted due to clogging of the capillary tip.
