We purified the FLAG fusion hEAG1 channel protein from HEK-293T cells stably overexpressed hEAG1. The function of this fusion protein has been demonstrated by using the patch-clamp method and the quality and specificity of purified protein are confirmed by Western blot (Figure 1). The purified channel protein is biotinylated to perform an interaction assay with the lipids (PIP2) by using the real-time BLI assay. The BLI binding assay configuration is shown in Figure 2. A typical binding curve between hEAG1 and PIP2 is shown in Figure 3. In this case, 3 μM PIP2 is dissolved in PBS buffer (the configuration of PIP2 is shown in Supplementary Figure 1), and the signal is analyzed using a double reference subtraction protocol to subtract the non-specific binding (the binding between sensor and PIP2), background (the interaction between biotinylated hEAG1 protein and PBS), and signal drift (the binding between sensor and PBS) caused by sensor variability. And the binding trace is globally fit and shown a well-fitting overlay (Supplementary Figure 2). Also, we measure the kinetics of binding of PIP2 to the purified hEAG1 channel complex by incubating the proteins at different concentrations of PIP2. After analysis, we get a dissociation constant (Kd) value of 0.35 ±0.04 μM, which is similar to the IC50 value obtained from the electrophysiological measurements15. These results demonstrated that the BLI assay is appropriate for ion channel membrane protein and lipids interaction analysis.
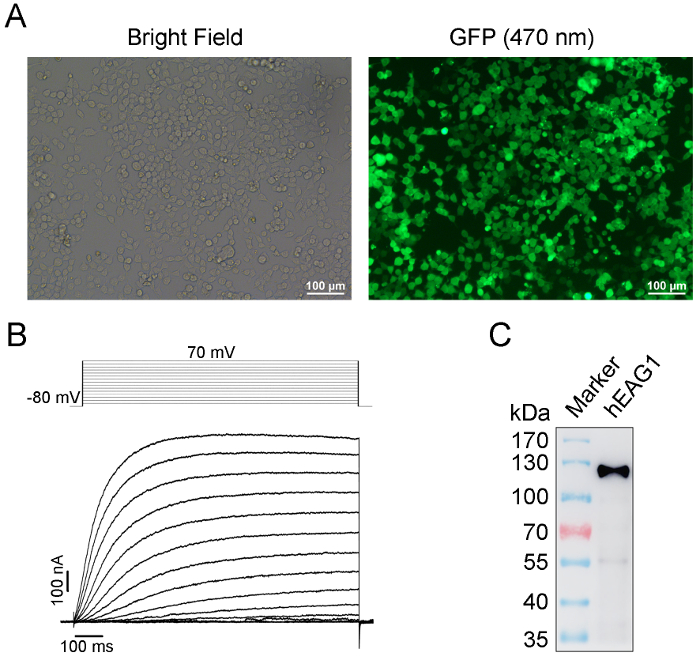
Figure 1: Identification of the expression, function, and specificity of recombinant hEAG1 protein from HEK-293T cells by GFP imaging, patch clamp, and western blot, respectively. (A) The stable expression hEAG1 HEK-293T system is successfully established by monoclonal puromycin-resistant selection after transfection with hEAG1-pCDH lentivirus system as evidenced by GFP expression in almost all cells. (B) Pulse protocol (top) and superimposed current traces from a representative whole-cell patch-clamp recording from hEAG1 channels in a stable cell in A. The current is elicited by depolarizing voltages from the holding voltage of -80 mV to 70 mV with the step of 10 mV followed by repolarization to -80 mV. The cells are incubated in the normal K+ channel recording solutions as described previously15. The voltage-dependent outward potassium currents suggest that functional hEAG1 channels are highly expressed in HEK293T cells. (C) Western blot of hEAG1 channel protein from purified protein samples. The anti-FLAG antibody recognizes a single protein band of ~110 kDa, demonstrating a full-length of FLAG-tagged hEAG1 channel expression. This Figure 1C has been modified from Han et al.15. Please click here to view a larger version of this figure.
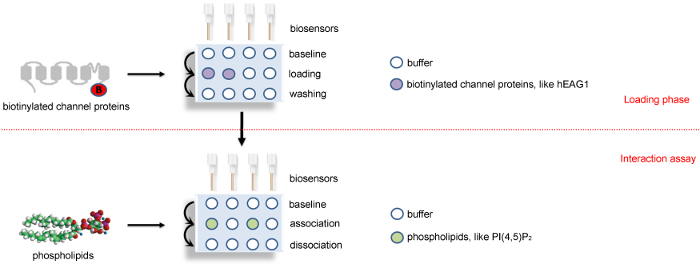
Figure 2: A schematic diagram showing the BLI binding assay protocol. Four sensors are used in parallel in biotinylated-channel proteins or their dissolved SD buffer to load the channel proteins and the references. After that, these four sensors are transferred to assay phase to detect the association and disassociation with phospholipids or its solution buffer. The positions of channel proteins, phospholipids, and buffer are colored as indicated. The horizontal red dotted line indicates the two major steps of BLI study: loading phase and interaction assay phase. This Figure 2 has been modified from Han et al.15. Please click here to view a larger version of this figure.
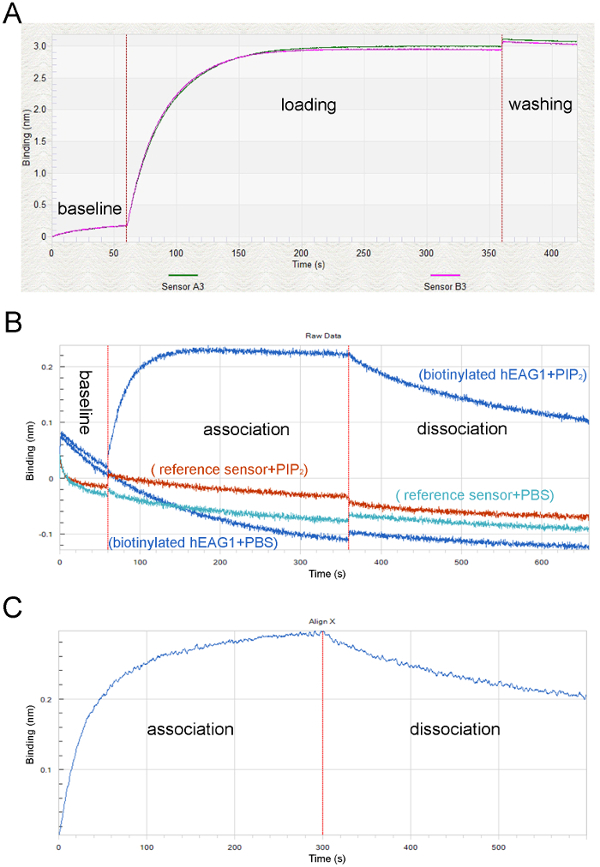
Figure 3: Screen captures showing the raw data and processed data in a typical BLI study. (A) Typical loading and equilibration curves showing the equilibration step (60 s) with SD-buffer (baseline), the loading step with hEAG1 proteins (loading) and the reference curve equilibrated and loaded with hEAG1 proteins (loading), simultaneous measurement of two individual sensor tips. (B) The vertical red lines indicate the transferring of sensors from the lipid solution to the buffer solution during assay operation. (C) The original optical signals at association and dissociation phases after processing the double reference subtraction to subtract the non-specific binding signals. Please click here to view a larger version of this figure.
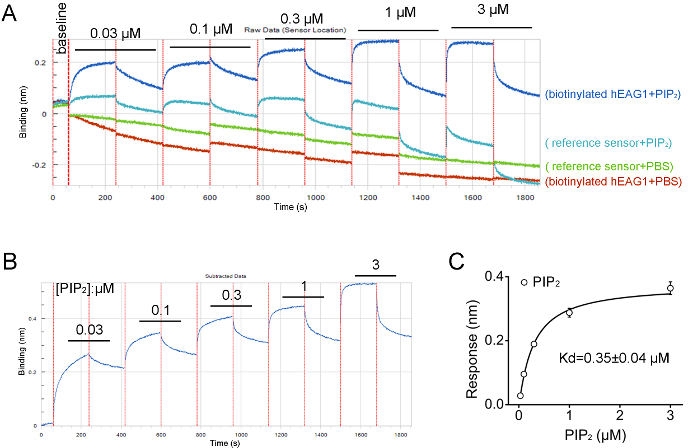
Figure 4: Results from BLI assay showing hEAG1 channel protein directly interacting with PIP2. (A) Screen capture showing the raw data of hEAG1 protein binding with PIP2 at concentration-dependence manner (0.03–3 μM). The accumulated concentrations of PIP2 denoted corresponding to the biosensors' data traces. (B) The raw data processing showing the changes in optical interference in different concentrations of PIP2 in a representative assay. (C) Curve fit with Hill equation obtained from the peak value of the optical interference signal measured at different PIP2 concentrations for determination of the equilibrium dissociation constants (Kd) of the interaction between the hEAG1 channel protein and PIP2 (n = 3).The Figure 4B and 4C have been modified from Han et al.15. Please click here to view a larger version of this figure.
Supplementary Figure 1: The configuration of long-chain phosphatidylinositol 4, 5-bisphosphate (PIP2). Please click here to download this figure.
Supplementary Figure 2: Screen capture of the fitted overlay from the BLI assay of Figure 3. The data is processed and fitted and only shown Association and Dissociation phases. The processed data curve is blue and the nonlinear fitting curve is red. Goodness of fit: R2 = 0.984789, X2 = 0.019109. The maximal binding parameter (Rmax) = 0.2875 nm (± 0.0006). Please click here to download this figure.
