The goal of this experiment was the deletion of Pim1 in MEL cells. Use of multiple non-overlapping sgRNA pairs (i.e., independent protospacer sequences) may help to control for off-target effects (Figure 1A). A consistent phenotype would be more likely to result from an on-target effect as opposed to a common off-target effect shared by multiple independent protospacer sequences. Each pair would lead to production of a unique deletion breakpoint. If close together (i.e., n less than ~150 bp), the same screening primers could be used to detect deletions produced by each set of sgRNAs. Genomic deletions may disrupt genes by using sgRNA pairs in various locations with respect to the gene (Figure 1B). For example, the sgRNA pair may flank a gene for deletion of the entire gene body; the pair could be located within two exons, with the potential to create frameshift indels even if one or both alleles were not deleted; or the pair may flank a specific exon to allow disruption of a particular isoform.
The deletion strategy used for Pim1 was to design flanking sgRNAs to delete the entire gene body, an 8 kb deletion (Figure 2). This strategy was chosen in part due to the relatively small size of the Pim1 gene. This example shows one PAM (green) on the top (Watson) strand and one PAM on the bottom (Crick) strand; however, DSB is independent of PAM sequence localization to the top or bottom strand. sgRNA pairs can have both PAM sequences on the top strand, both on the bottom strand, or one of each. Using the protocol described above, two sgRNA were designed, cloned into the pX330 expression vector, and delivered to MEL cells by electroporation along with a GFP reporter (Figure 2A). The top 3% of GFP+ cells were sorted two days post-electroporation and plated clonally at limiting dilution. Screening primers were designed as described in step 2 and as shown in Figure 2. PCR conditions were optimized using gDNA isolated from parental MEL cells and from “bulk” sorted cells.
10 days after plating, gDNA was isolated from all clones and screened for deletion via PCR, which identified non-deletion, monoallelic and biallelic deletion clones according to the patterns of non-deletion (ND) and deletion (D) amplicons (Figure 3). Non-deletion clones were identified as having the presence of the non-deletion amplicon and the absence of the deletion amplicon. Monoallelic clones were identified as having the presence of both the non-deletion and deletion amplicons. Biallelic clones were identified as having the absence of the non-deletion amplicon and the presence of the deletion amplicon. For this deletion, 400 clones were screened which identified 126 monoallelic deletion clones and 32 biallelic deletion clones (it is important to note that deletion frequency varies with deletion size12). Biallelic deletion clones were selected and moved to flasks with 8 ml of media. After allowing 5 days for expansion, each clone was retested by PCR of gDNA to confirm biallelic deletion and deletion amplicons were subjected to Sanger sequencing to identify the precise deletion (Figure 2B). Heterogeneity within the deletion amplicons reflects imperfect indel-forming NHEJ repair. Sequencing of the non-deletion allele in monoallelelic deletion clones uncovered indels in the majority of cases, demonstrating that even the non-deleted allele is frequently edited by CRISPR/Cas9, which may be important for applications where both monoallelic and biallelic deletion clones are required. RNA was isolated from biallelic deletion clones and analyzed by RT-qPCR to confirm loss of Pim1 expression (Figure 4).
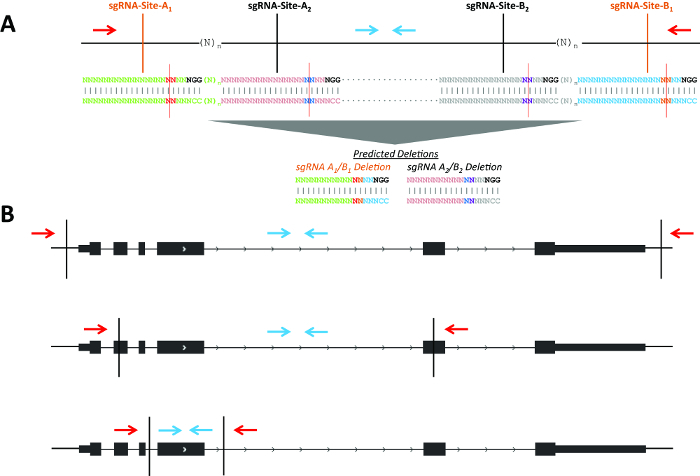
Figure 1. Schematic of possible deletion strategies. (A) Two example sgRNA pairs for genomic deletions (shown in black and orange, respectively). The blue arrows indicate primers to detect the non-deletion amplicon and red arrows indicate primers to detect the deletion amplicon. sgRNA positions 17 and 18 are highlighted in red and blue at sgRNA-Sites-A1/A2 and are highlighted in purple and orange at sgRNA-Sites-B1/B2 with a red line indicating the predicted Cas9 cleavage between positions 17 and 18. (B) CRISPR-directed cleavages are shown as vertical black lines. The blue arrows indicate primers to detect the non-deletion amplicon and red arrows indicate primers to detect the deletion amplicon. Please click here to view a larger version of this figure.
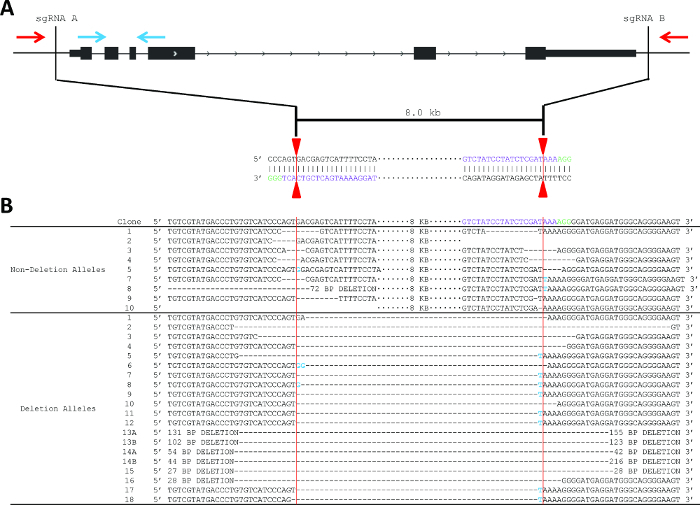
Figure 2. Strategy to produce and detect deletion of Pim1, including sequencingdeletion and non-deletion alleles. (A) Schematic of the PCR-based screening strategy to identify Pim1 deletion clones. One primer pair is located internal to the deletion (blue arrows) and one primer pair is localized external to the deletion (red arrows). sgRNA sequences (protospacer sequences) are shown in purple. The vertical red lines indicate the predicted Cas9 cleavage between positions 17 and 18 of the sgRNA sequence. (B) Sanger sequencing reveals indel formation at the sgRNA recognition site. sgRNA sequences are shown in purple and PAM sequences in green. Deletion events are shown by an equivalent number of dash marks and insertions are highlighted in blue. Vertical red lines indicate predicted cleavage site, between positions 17 and 18 of the sgRNA. Please click here to view a larger version of this figure.
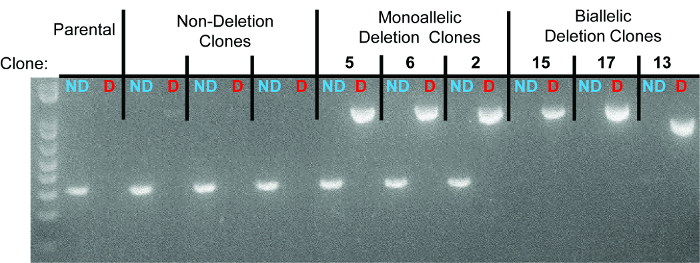
Figure 3. Representative gel identifying non-deletion, monoallelic, and biallelic clones. For each clone, the left lane represents the non-deletion amplicon (“ND” in blue) and the right lane represents the deletion amplicon (“D” in red). Individual clones are separated by dotted lines. The three monoallelic deletion clones are clones 5, 6, and 2. The three biallelic deletion clones are clones 15, 17, and 13. As seen in the sequencing data, the deletion band for clone 13 has a smaller size due to larger deletions at the deletion junction.
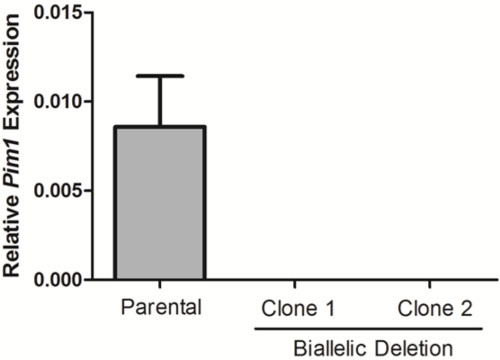
Figure 4. Loss of Pim1 expression in biallelic deletion clones. Pim1 expression was calculated for two biallelic deletion clones by RT-qPCR. Data was normalized to Gapdh using the 2–ΔCt method.
| Protospacer Sequence | |
| sgRNA-A | 5’-TAGGAAAATGACTCGTCACT-3’ |
| sgRNA-B | 5’-GTCTATCCTATCTCGATAAA-3’ |
Table 1. 20-mer protospacer sequences for two sgRNA for the deletion of Pim1.
| Reverse Complement of Protospacer Sequence | |
| sgRNA-A-rc | 5’-AGTGACGAGTCATTTTCCTA-3’ |
| sgRNA-B-rc | 5’-TTTATCGAGATAGGATAGAC-3’ |
Table 2. Reverse complement of the protospacer sequences for the two sgRNA from Table 1.
| Sequences | |
| sgRNA-A | 5’-CACCTAGGAAAATGACTCGTCACT-3’ |
| sgRNA-A-rc | 5’-AAACAGTGACGAGTCATTTTCCTA-3’ |
| sgRNA-B | 5’-CACCGTCTATCCTATCTCGATAAA-3’ |
| sgRNA-B-rc | 5’-AAACTTTATCGAGATAGGATAGAC-3’ |
Table 3. Protospacer sequences and their reverse complements with “CACC” and “AAAC” added for cloning into the pX330 vector using BbsI restriction enzyme.
| Sequences | |
| sgRNA-A | 5’- CACCGTAGGAAAATGACTCGTCACT-3’ |
| sgRNA-A-rc | 5’- AAACAGTGACGAGTCATTTTCCTAC-3’ |
| sgRNA-B | 5’- CACCGTCTATCCTATCTCGATAAA-3’ |
| sgRNA-B-rc | 5’- AAACTTTATCGAGATAGGATAGAC-3’ |
Table 4. sgRNA expression from the U6 promoter of the pX330 vector is enhanced by the addition of a G nucleotide after the CACC sequence and before the 20-mer. The addition of an extra G nucleotide requires the addition of a C nucleotide at the 3’ end of the reverse complement oligo (e.g., sgRNA-A). However, if the first position of the 20-mer (protospacer sequence) is already a G nucleotide, there is no need to add another G (e.g., sgRNA-B) and no need to add C to the final position of the reverse complement oligo.
