1. Isolate total RNA
- Add 1ml Trizol to cells/tissue, and homogenize with 18-22 gauze needle if necessary.
- Add 200 μl chloroform, and spin at 14,000 rpm for 30 minutes at 4 °C.
- Take out top aqueous layer, add 0.5 μl linear acrylamide, then add 500 μl isopropanol. Let sit at room temperature for 20 minutes.
- Spin at 14,000 rpm at 4 °C, wash pellet with 70% ethanol, and vacuum dry.
- Resuspend in 1 μl nuclease free water and transfer to 200 μl PCR tube.
2. Prepare double stranded cDNA
- To the above 1 μl RNA add 1 μl of 100 μM oligo-dT-T7 reverse transcription primer (5′- GGCCAGTGAATTGTAATACGACTCACTATAGGGAGGCGGTTTTTTTTTTTTTTTTTTTTTTTT -3′), heat at 70 °C (heat block) for 5 min, and snap cool on ice.
- Add 1 μl 5x FS buffer, 0.5 μl DTT, 0.5 μl dNTP mix and 0.5 μl RnaseOUT (from SuperScript II kit).
- Heat to 42 °C in PCR machine for 1 minute, then add 0.5 μl Superscript II reverse transcriptase. Incubate for 1 hr at 42 °C.
- Heat at 70 °C for 10 minutes, cool to 4 °C.
- On ice, add the following to the above reaction:
- Nuclease free water – 22.75 μl
- 5X Second strand buffer – 7.5 μl
- dNTP mix – 0.75 μl
- E. coli DNA Ligase – 0.25 μl
- E. coli DNA polymerase – 1 μl
- RnaseH – 0.25 μl
Incubate for 2 hr at 16 °C then add 1 μl T4 DNA Polymerase, and incubate for additional 10 minutes at 16 °C.
- Transfer to 1.5 ml eppendorf tube, and add 80 μl water.
- Add 0.5 μl of linear acrylamide.
- Add 72 μl 3 M NH4OAc and 480 μl of cold 100% ethanol. Precipitate for 1 hr at -20 °C.
- Spin at 14,000 rpm at 4 °C for 30 minutes, wash with 1 mL 70% ethanol, centrifuge at 14,000 rpm for 2 minutes, and vacuum dry for about 10 minutes.
3. Amplify poly-A mRNA by in vitro transcription
- Resuspend above cDNA in 3.5 μl nuclease free water.
- To the above, add 1 μl each of the dNTPs (total 4 μl), 1 μl of 10X reaction buffer and 1 μl of T7 polymerase, and 0.5 μl of RnaseOUT from Megascript kit.
- Incubate at 37 °C in PCR machine overnight.
- Ready for next step. Can be stored at -80 °C.
4. Fragmentation of amplified poly-A mRNA
- Add 26 μl of water to above reaction and 4 μl 10x fragmentation reagent.
- Heat in PCR machine at 70 °C for exactly 7 minutes.
- Add 5 μl of fragmentation stop buffer, put sample on ice.
5. RNA clean up
- To the above reaction, add 60 μl water and 350 μl of buffer RLT from Rneasy MinElute kit, and mix by pipetting.
- Add 250 μl ethanol, mix by pipetting, and pipette into spin column.
- Spin at 8000 rcf for 20 seconds.
- Wash once with RPE, spin for 20 seconds, wash second time with 80% EtOH, spin for 2 minutes, and dry column with 5 minute spin.
- Elute RNA in 10 μl nuclease free water.
6. cDNA synthesis
First strand synthesis for single read library:
- In a PCR tube, add the following:
- Fragmented Poly-A mRNA – 10 μl
- NotI Random nonamer Primer – 1 μl
Incubate at 70 °C for 5 minutes in thermocycler, and quick chill on ice. NotI Nonamer Primer (5′- TGAATTCGCGGCCGCTCAAGCAGAAGACGGCATACGAGCTCTTCCGATCT NNNNNNNNN -3′). The 5′ proximal sequence is the NotI restriction site while the next sequence until the random region is the reverse complement of Illumina’s adaptor B sequence from Chip-Seq kit.
- To the above, add the following on ice (from SuperScript III kit):
- 5X first strand buffer -4 μl
- DTT – 2 μl
- dNTP mixture* – 1.5 μl
Put on thermocycler for 2 minutes at 42°C, add 1 μl of SuperScript III reverse transcriptase, and incubate at 42 °C for 1hr.
*Instead of dCTP, 5-methyl dCTP was used in the dNTP mixture.
First strand synthesis for paired end library:
- In a PCR tube, add the following:
- Fragmented Poly-A mRNA – 10 μl
- Random hexamer primer – 1 μl
Incubate at 65 °C for 5 minutes in thermocycler, and quick chill on ice.
- To the above, add the following on ice (from SuperScript firststrand III kit):
- 5X first strand buffer – 4 μl
- DTT – 2 μl
- dNTPs (from kit) – 1.5 μl
Put on thermocycler for 1 min at 45 °C, add 1 μl of SuperScript III reverse transcriptase, and incubate at 45 °C for 1 hour.
Second strand synthesis (for both libraries):
- To the above, add the following on ice (from SuperScript II kit):
- Water (RNase free) – 91 μl
- 5X Second Strand Buffer – 30 μl
- dNTPs (from kit) – 3 μl
- E. coli DNA Ligase – 1 μl
- E. coli DNA polymerase – 4 μl
- E. coli RNase H – 1 μl
Mix tube by inversion, give a short spin and incubate at 16 °C for 2 hr.
7. Purify cDNA
- Purify cDNA sample with Zymo columns, elute in 40 μl of water for single read and 30 μl of water for paired end library.
8. End repair
For single read library:
(Use Illumina Chip-Seq sample prep kit)
- To the above 40 μl of cDNA, add:
- T4 DNA ligase buffer with 10mM ATP – 5 μl
- dNTP mix – 2 μl
- T4 DNA polymerase – 1 μl
- Klenow enzyme (diluted 1:5 with water to 1U/ μl) – 1 μl
- T4 PNK – 1 μl
Incubate in the thermal cycler for 30 minutes at 20 °C.
For paired end library:
(Use Illumina paired end sample prep kit)
- To the above 30 μl cDNA, add:
- RNase DNase free water – 45 μl
- T4 DNA ligase buffer with 10mM ATP – 10 μl
- 10mM dNTP mix – 4 μl
- T4 DNA polymerase – 5 μl
- Klenow enzyme – 1 μl
- T4 PNK – 5 μl
Incubate in the thermal cycler for 30 minutes at 20 °C.
9. cDNA clean up
- Clean up cDNA using Zymo columns. Elute in 34 μl of EB for single read and 32 μl of EB for paired end library.
10. Add ‘A’ bases to the 3′ end of the DNA fragments
For single read library:
- Prepare the following reaction mix:
- DNA sample – 34 μl
- Klenow buffer – 5 μl
- dATP – 10 μl
- Klenow exo – 1 μl
Incubate for 30 minutes at 37 °C.
For paired end library:
- Prepare the following reaction mix:
- DNA sample – 32 μl
- Klenow buffer – 5 μl
- dATP – 10 μl
- Klenow exo – 3 μl
Incubate for 30 minutes at 37 °C.
11. cDNA clean up
- Clean up cDNA using zymo columns. Elute in 10 μl of EB.
12. Adapter ligation
For single read library:
(Use Illumina Chip-Seq sample prep kit)
- Prepare the following reaction mix:
- DNA sample – 10 μl
- Adapter Oligo Mix – 1 μl
- 2X DNA Ligase Buffer- -15 μl
- DNA ligase – 4 μl
Incubate for 15 minutes at room temperature. Adapters should be thawed on ice and diluted 1:20.
For paired end library:
(Use Illumina paired end sample prep kit)
- Prepare the following reaction mix:
- DNA sample – 10 μl
- PE Adapter Oligo Mix – 10 μl
- 2X DNA Ligase Buffer – 25 μl
- DNA ligase – 5 μl
Incubate for 15 minutes at 20 °C. Adapters should be thawed on ice and diluted 1:20.
13. Ligation reaction clean up
- Clean up the ligation reaction using zymo columns. Elute in 44 μl of water for single read library and 6 μl of EB followed by another elution in 5 μl of EB for paired end library.
Perform a NotI digestion, ONLY for single read library
- cDNA – 44 μl
- NEB buffer 3 – 5 μl
- BSA – 0.5 μl
- NotI – 1 μl
Incubate for 2 hr to overnight at 37 °C, and purify reaction using zymo column. Elute with 6 μl of buffer EB followed by a second elution with 5 μl of EB.
14. Size selection/Gel purification
Perform the following using a 2% Sybr Safe E-Gel from Invitrogen.
- Run Program 0-PreRun 2 minutes.
- Load 1kb plus ladder from Invitrogen diluted 1:4, and load 10 μl.
- Load all of DNA sample, and fill empty lanes with 10 μl of water.
- Run Program 1-EGel 2% Run 28 minutes.
- Size select 200-300 bp gel slice with a fresh razor blade.
15. Elute DNA from gel slice
- Weigh gel slice, and add 3 volumes of buffer QG to 1 volume of gel (use Gel Extraction kit from Qiagen)
- Incubate at 50 °C for 10 minutes or until gel slice dissolves.
- Add 1 volume of ispropanol, and mix by inversion or pipetting.
- Add to column, spin for 1 minute at maximum speed, and discard flow-through.
- Add 500 μl of Buffer QG to column, spin for 1 minute at maximum speed, and discard flow through.
- Add 750 μl of Buffer PE to column, spin for 1 minute at maximum speed, and discard flow through.
- Centrifuge for 1 minute at maximum speed.
- Elute in 36 μl of EB for single read and 23 μl EB for paired end library.
16. PCR
For single read library:
(Use Illumina Chip-Seq sample prep kit)
- Prepare the following PCR reaction mix:
- DNA – 36 μl
- 5X Phusion buffer – 10 μl
- dNTP mix – 1.5 μl
- PCR primer 1.1 – 1 μl
- PCR primer 2.1 – 1 μl
- Phusion polymerase – 0.5 μl
Use the following PCR protocol:
- 30 seconds at 98 °C
- 10 cycles of:
- 10 seconds at 98 °C
- 30 seconds at 65 °C
- 30 seconds at 72 °C
- 5 minutes at 72 °C
- Hold at 4°C
*The first time that the kit is used, dilute PCR primers 1:2 with EB buffer.
For paired end library:
(Use Illumina paired end sample prep kit)
- Prepare the following PCR reaction:
- DNA – 23 μl
- Phusion DNA polymerase – 25 μl
- PCR primer PE 1.1 – 1 μl
- PCR primer PE 2.1 – 1 μl
Amplify using the following PCR protocol:
- 30 seconds at 98°C
- 10 cycles of:
- 10 seconds at 98°C
- 30 seconds at 65°C
- 30 seconds at 72°C
- 5 minutes at 72°C
- Hold at 4°C
*The first time that the kit is used, dilute PCR primers 1:2 with EB buffer.
17. Library clean up
- Clean up library using zymo columns. Elute in 12 μl of EB
18. Quantitate library
- Quantitate library using Qubit. It is ready for sequencing. Use sequencing primers from Illumina single read cluster generation kit V4 or higher for single read library and Illumina paired end cluster generation kit V4 or higher for paired end library.
19. Data analysis
Bowtie6 was used to map reads to the RefSeq gene set (NCBI Build 36.1). The single end reads (30 nucleotides) and the pair end reads (42 nucleotides) were mapped allowing up to 10 matches to the gene set, and allowing up to two mismatches per read. Transcripts Per Million (TPM) values were obtained to measure gene expression using RSEM7 (RNA-Seq by Expectation-Maximization).
20. Representative Results: We made T7LA libraries for both single read and paired end runs from 1 μg, 100 μg, 10 μg, 1 μg and 100 pg starting total RNA (Figure 1). For evaluation of our protocol, we made single read and paired end libraries without T7 RNA amplification starting from 10 μg total RNA These control libraries, termed “MinAmp”, have minimal amplification. The only amplification they undergo are the 10 cycles of PCR near the end of the protocol to ligate the Illumina sequencing adapters, a step common to all libraries. All RNA used were isolated from H14 human embryonic stem cells8.
We first evaluated the number of genes identified by the various libraries (Table 1 and Supplementary Table 1). For both single read and paired end libraries, the 10 ng T7LA libraries identified almost the same number of genes as the 10 μg MinAmp libraries, with a TPM of 10 or more. In the case of the single read libraries, the 10 ng T7LA library identified 100% of the 8500 genes identified by the 10 μg unamplified library. For paired end libraries, the 10 ng T7LA library identified 86% of genes identified by the 10 μg unamplified library (7961 of 9267 genes). Libraries made from less than 10 ng were not able to identify as many genes. For example, in the single read protocol, the 1 ng library identified only ˜50% of the genes identified by the 10 μg MinAmp library, prompting us to limit the lowest amount of total RNA for use with the T7LA protocol to 10 ng. Moreover, mapping of a housekeeping gene, GAPDH (Figure 2) shows that all T7LA libraries made with at least 10ng of starting RNA identified all exons, including the extreme 5′ exon. Comparison of the 10 ng T7LA single read and paired end libraries with the MinAmp libraries shows a high degree of similarity (Spearman correlation, R = 0.90 and 0.95 respectively, Figures 3a and b). We also compared the two single read and paired end libraries made from 10 ng total RNA and they had a very high correlation coefficient (R = 0.92), demonstrating that both types of libraries made using the T7LA protocol produce a very similar gene expression signature (Figure 3c.). Hence, the T7LA method is able to produce sequencing libraries that are as reliable and comprehensive as the MinAmp libraries, but from 1000-fold less starting material.
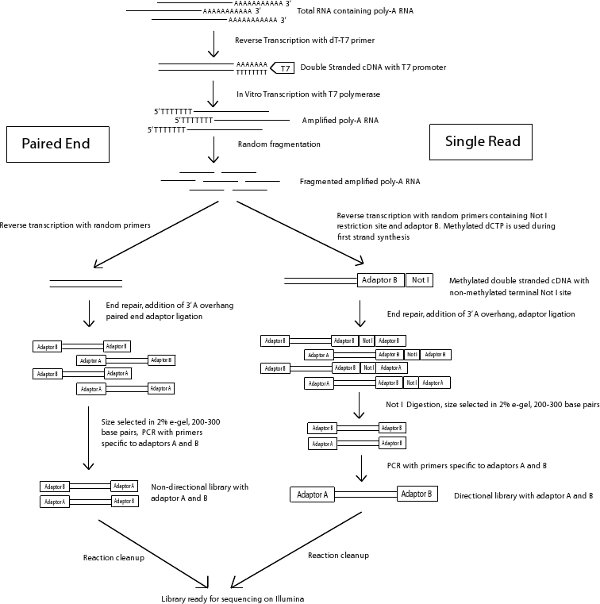
Figure 1 Schema of paired end and single read library preparation protocol.
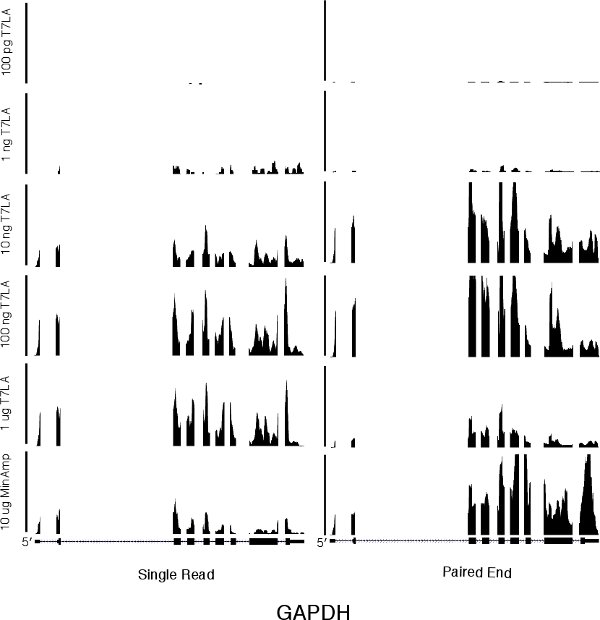
Figure 2 A genome browser picture of a housekeeping gene, GAPDH, for all single read and paired end libraries. The scale bar on the left for the single read libraries indicates 350 total reads. The scale bar in the center for the paired end libraries indicates 5000 total reads. The horizontal axis represents the genome sequence of GAPDH.
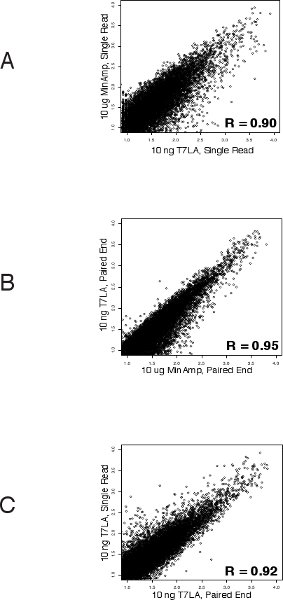
Figure 3 Correlation of gene expressions between the various libraries (Spearman’s): A. Between single read 10 ng T7LA and 10 μg MinAmp library shows that both these libraries have a very similar gene expression pattern (R = 0.90). B. Between paired end 10 ng T7LA and 10 μg MinAmp library demonstrates their similarity of gene expression profiles (R = 0.95). C. Correlation of gene expressions between 10 ng paired end and 10 ng single read libraries show a high degree of similarity between these libraries prepared by the T7LA method (R = 0.92).
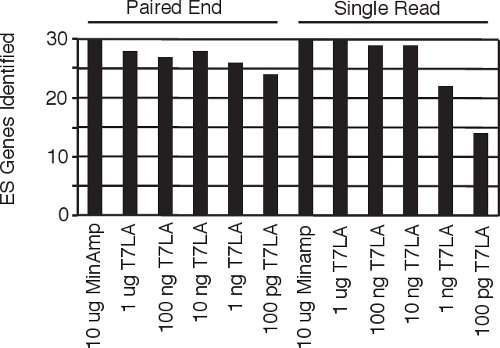
Figure 4 Identification of human embryonic stem cell specific genes5 from all the single read and paired end libraries.
| Library | Type° | Raw Clusters | % Clusters passing filter | % Aligning to genome | % Error Rate | Genes identified |
| MinAmp 10ug | Single read | 225602 +/- 4952 | 65.48 +/- 2.58 | 47.61 +/- 0.53 | 0.62 +/- 0.06 | 8500 |
| 1ug T7LA | Single read | 144818 +/- 6513 | 82.21 +/- 6.45 | 48.09 +/- 0.27 | 0.42 +/- 0.03 | 8757 |
| 100ng T7LA | Single read | 27385 +/- 1818 | 81.33 +/- 11.75 | 44.46 +/- 4.53 | 0.49 +/- 0.10 | 8709 |
| 10ng T7LA | Single read | 11184 +/- 985 | 60.70 +/- 3.70 | 14.96 +/- 1.15 | 0.99 +/- 0.30 | 8589 |
| 1ng T7LA | Single read | 12695 +/- 1365 | 53.27 +/- 16.76 | 4.08 +/- 0.79 | 2.25 +/- 1.56 | 4720 |
| 100pg T7LA | Single read | 10390 +/- 1398 | 72.99 +/- 2.90 | 1.48 +/- 0.20 | 1.51 +/- 0.39 | 1121 |
| MinAmp 10ug | Paired End R1 | 95786 +/- 12937 | 90.77 +/- 2.79 | 58.50 +/- 0.95 | 0.94 +/- 0.38 | 9267 |
| Paired End R2 | 95786 +/- 12937 | 90.77 +/- 2.79 | 58.13 +/- 1.13 | 0.99 +/- 0.37 | ||
| 1ug T7LA | Paired End R1 | 297669 +/- 10196 | 91.35 +/- 0.36 | 46.89 +/- 0.14 | 0.47 +/- 0.01 | 7334 |
| Paired End R2 | 297669 +/- 10196 | 91.35 +/- 0.36 | 45.52 +/- 0.12 | 0.51 +/- 0.01 | ||
| 100ng T7LA | Paired End R1 | 205602 +/- 9932 | 90.53 +/- 0.76 | 63.44 +/- 1.00 | 0.48 +/- 0.02 | 8011 |
| Paired End R2 | 205602 +/- 9932 | 90.53 +/- 0.76 | 61.80 +/- 8.09 | 0.60 +/- 0.36 | ||
| 10ng T7LA | Paired End R1 | 214622 +/- 11155 | 89.98 +/- 1.13 | 56.32 +/- 1.94 | 0.80 +/- 0.26 | 7961 |
| Paired End R2 | 214622 +/- 11155 | 89.98 +/- 1.13 | 46.41 +/- 18.39 | 2.48 +/- 2.68 | ; | |
| 1ng T7LA | Paired End R1 | 144951 +/- 19841 | 90.54 +/- 1.19 | 3.91 +/- 0.16 | 8.71 +/- 0.86 | 8124 |
| Paired End R2 | 144951 +/- 19841 | 90.54 +/- 1.19 | 3.27 +/- 1.21 | 9.11 +/- 3.52 | ||
| 100pg T7LA | Paired End R1 | 187600 +/- 11759 | 89.52 +/- 1.11 | 1.78 +/- 0.05 | 13.42 +/- 0.50 | 6623 |
| Paired End R2 | 187600 +/- 11759 | 89.52 +/- 1.11 | 1.99 +/- 0.23 | 15.29 +/- 0.96 | ||
° R1 and R2 are forward and reverse sequences of a tag
* ≥10 TPM
Table 1. Information on cluster numbers, genes identified, error rate, percent alignment of the single read and paired end libraries.
Supplementary Table 1. List of all genes and their TPM values for all samples, single read and paired end.
