Isolation and Culture of Primary Embryonic Mouse Midbrain Dopamine Neurons
Abstract
Source: Er, S., et al. Studying Pre-formed Fibril Induced α-Synuclein Accumulation in Primary Embryonic Mouse Midbrain Dopamine Neurons. J. Vis. Exp. (2020).
The video demonstrates the process of isolating and culturing dopamine neurons from the midbrain of mouse embryos. The isolated ventral midbrain floors are dissociated using enzymes. The resulting dopamine neurons are harvested and transferred into micro-islands inside multi-well plates. The cells are incubated with a dopamine neuron medium to maintain the cells at a high density inside the micro-islands.
Protocol
All procedures involving sample collection have been performed in accordance with the institute's IRB guidelines.
1. Preparation
- Prepare dopamine neuron medium (DPM) with 0.46% D-glucose, 1% L-glutamine, 1% N2, 0.2% primocin, completed with Dulbecco's modified Eagle medium/nutrient mixture F-12 (DMEM/F12). Filter the DPM after mixing the ingredients. Store DPM at 4 °C and warm each aliquot only once.
NOTE: DPM should not contain Glial cell line-derived growth factor (GDNF), as it will reduce α-synuclein accumulation in dopamine neurons. - Prepare siliconized glass pipettes that are extremely hydrophobic, thereby minimizing the attachment to the surface and loss of cells during the initial handling of embryonic neurons.
- Add 10 mL of siliconizing fluid to 1 L of distilled water and mix by stirring in a 2 L vessel. Leave the glass pipettes immersed in the siliconizing solution for 15 min.
- Rinse the pipettes 3–5x with distilled water. Dry the pipettes overnight at room temperature (RT) or for 1– 2 h at 100-120 °C heated sterile space to speed up the drying.
- Sterilize the pipettes by standard autoclaving in a sealed autoclave bag.
- Prepare poly-L-ornithine (PO) coated 96 well plates with transparent bottoms by adding 60 μL of PO solution into the middle wells of the 96 well plate to be used for seeding of the neurons, leaving at least one row/column of wells at the edges of the plate to avoid edge effects. Keep the coated plate overnight at 4 °C or 4 h at RT.
- Prior to plating the cells, aspirate PO completely and wash the cells thrice with 100 μL of 1x phosphate-buffered saline (PBS). Aspirate 1x PBS from the wells and keep the lid of the plate open for complete drying.
NOTE: It is possible to collect used PO and filter it for reusing. This can be repeated twice for the same PO solution. - Add 50 μL of DPM to previously coated wells. Aspirate DPM from the wells with a 100 μL plastic tip and simultaneously scratch the bottom of the well with circular movements to remove the coating at the perimeter of each well. A PO-coated island will remain in the middle of the well.
- Under a laminar hood, add 10 μL of DPM to the middle of each coated island to create micro islands.
NOTE: A plate with DPM-covered micro islands can be kept under the laminar flow hood for 1–2 h during the isolation of cells
2. Isolation of the ventral midbrain floor from embryonic day 13.5 (E13.5) mouse embryos
- Prior to dissection, fill a 10 cm Petri dish with Dulbecco's buffer and keep it on ice.
- Euthanize a E13.5 pregnant female mouse according to the institution's guidelines. Place the mouse flat on its back and spray the anterior body with 70% ethanol. Lift the skin above the womb with forceps and make an incision with surgical scissors to expose the uterus.
- Carefully remove the uterus and place it into the previously prepared Petri dish on ice.
- Using surgical scissors under the laminar hood at RT, carefully remove the embryos from the uterus. Remove all placental residue from the embryos with forceps and place them into a new 10 cm Petri dish filled with Dulbecco's buffer.
- Using dissection forceps or needles, cut off the hindquarter of the head from the places marked with black arrows in Figure 1A. Take the cut piece away from the rest of the embryo (Figure 1B).
- Place the posterior of the cut piece towards the observer (Figure 1C) and gently cut it open from caudal to cranial (Figure 1D). From 0.5 mm below the cranial opening, cut a 2 mm2 –3 mm2 region, shown in Figure 1E.
- Collect the ventral midbrain floor (see Figure 1F) in an empty 1.5 mL microcentrifuge tube. Keep the microcentrifuge tube on ice until all midbrain floors are collected in it.
NOTE: Alternatively, the midbrain floors can be collected with a 1 mL micropipette after dissection of all embryo brains.
3. Establishing primary embryonic midbrain cultures from E13.5 mouse embryos in 96 well plate format
- After the collection of midbrain floors from all embryos in the same 1.5 mL tube, remove the residual Dulbecco's buffer and wash the tissue pieces thrice with 500 μL of Ca2+, Mg2+-free Hank's Balanced Salt Solution (HBSS).
- Remove HBSS and add 500 μL of 0.5% trypsin to the tube. Incubate it at 37 °C for 30 min.
- During incubation, warm 1.5 mL of fetal bovine serum (FBS) at 37 °C, add 30 μL of DNase I to the FBS, and mix. Also, fire-polish the tip of a siliconized glass pipette. Make sure that the hole has no sharp edges and is around the same size as a 1 mL micropipette tip.
NOTE: As an alternative, a low adhesion 1 mL micropipette tip can be used for trituration. However, siliconized glass pipettes seem to give the best results. - As soon as the incubation in step 3.2 ends, add 500 μL of the FBS/DNase mix to the partially digested tissue. Use the glass pipette to triturate the tissue in the FBS/ trypsin mix. Triturate until tissues dissociate into tiny, barely visible particles. Avoid bubbles during trituration.
- Let the leftover particles precipitate at the bottom of the microcentrifuge tube by gravity. Without pipetting the precipitate at the bottom, collect the supernatant into an empty 15 mL conical polypropylene tube.
- Dilute FBS/DNase I from step 3.3 (98:2) with 1,000 μL of HBSS to obtain FBS/DNase-I/HBSS (49:1:50). Mix by pipetting up and down. Add 1,000 μL of the new mix to the leftover particles in the microcentrifuge tube. Triturate again and repeat step 3.5.
- Repeat the previous step once more to use up all FBS/ DNase-I/HBSS (49:1:50).
- Once all the supernatant is collected inside the 15 mL tube (from steps 3.5, 3.7, and 3.8), use a tabletop centrifuge to spin down the supernatant (~3 mL) at 100 x g, for 5 min. Remove the supernatant without touching the pelleted cells at the bottom.
- Wash the cell pellet by adding 2 mL of DPM to the tube and spin it down at 100 x g for 5 min. Remove the supernatant and repeat the washing 2x to minimize the debris in the pelleted cells.
NOTE: Always use fresh, warmed DPM for the cultured neurons. For the washing steps, DPM does not have to be fresh, but should be prewarmed to 37 °C. - Dilute the cells with fresh, warm DPM and transfer them to a microcentrifuge tube. The amount of DPM for dilution depends on the number of embryos used for tissue dissection. For example, use 150 μL of DPM to dilute the cells obtained from ten embryos.
- Transfer 10 μL of cells in DPM to a microcentrifuge tube. Mix them with 10 μL of 0.4% Trypan blue stain. Count live (i.e, Trypan blue negative) cells using a hemocytometer or an automated cell counter.
NOTE: Use 30,000 cells for plating per well to obtain ~1,000 dopamine neurons per well. If the cell density is higher than ~30,000 cells per 6 μL, further dilute the cells with DPM before plating so that the seeding volume is no less than 6 μL. - Without touching the bottom of the wells, remove the DPM from the micro islands created at step 1.6.
- In order to obtain reproducible cell density at each well, mix the cells by gentle pipetting prior to plating in the well. With a 1–10 μL micropipette, add 6 μL of cells to the middle of the well, at the location of each former micro island.
- Fill the empty wells at the edges of the plate with 150 μL of water or 1x PBS to minimize evaporation from the wells containing neuronal cultures. Incubate the plate in an incubator at 37 °C, 5% CO2 for 1 h.
- After 1 h, remove the plate from the incubator, add 100 μL of DPM into each well with cells and place it back in the incubator.
- Two days after plating (day in vitro 2, or DIV2), remove 25 μL and add 75 μL of fresh DPM to bring the final media volume to 150 μL and avoid evaporation as much as possible.
- Exchange half of the medium with fresh DPM (i.e., remove 75 μL and add 75 μL fresh DPM) at DIV5. Do not perform any media changes after DIV5.
Representative Results
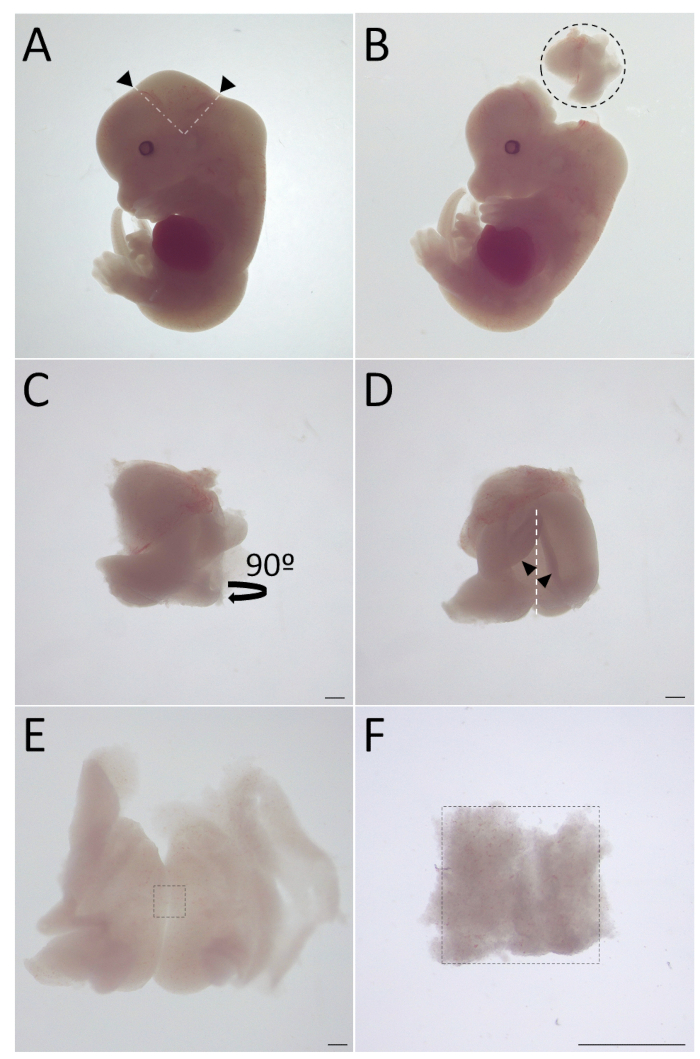
Figure 1: Dissection of midbrain floor from E13.5 mouse embryo. (A) Cutting locations at the hindquarter of the head is marked with black arrows and white dashed lines. (B) The piece was removed from the rest of the embryo. The removed piece is circled. (C) The piece was turned 90° to face the posterior towards the observer. (D) The piece was opened from the black arrows, from caudal to cranial (marked with white dashed line). (E) From 0.5 mm–1 mm below the opening, the 2 mm2 –4 mm2 region was cut (marked with black lines). (F) The ventral midbrain floor was isolated (marked with black dashed square). Scale bars = 1 mm.
Disclosures
The authors have nothing to disclose.
Materials
| 0.4% Trypan Blue stain | Gibco, Thermo Scientific, Waltham, Massachusetts, USA | 15250061 | |
| 15 ml CELLSTAR Polypropylene tube | Greiner Bio-One GmbH, Frickenhausen, Germany | 188261 | |
| 4',6-diamidino-2-phenylindole (DAPI) | Sigma-Aldrich, St. Louis, Missouri, USA | 10236276001 | |
| 5 M HCl | N/A | N/A | Media kitchen, Institute of Biotechnology, University of Helsinki |
| 5 M NaOH | N/A | N/A | Media kitchen, Institute of Biotechnology, University of Helsinki |
| 96-well ViewPlate (Black) | PerkinElmer, Waltham, Massachusetts, USA | 6005182 | |
| 99.5% Ethanol (EtOH) | Altia Oyj, Rajamäki, Finland | N/A | |
| AquaSil Siliconizing Fluid | Thermo Scientific, Waltham, Massachusetts, USA | TS-42799 | |
| Autoclaved 1.5 ml microcentrifuge tube | N/A | N/A | Media kitchen, Institute of Biotechnology, University of Helsinki |
| Bioruptor sonication device | Diagenode, Liege, Belgium | B01020001 | |
| Ca2+, Mg2+ free Hank's Balanced Salt Solution (HBSS) | Gibco, Thermo Scientific, Waltham, Massachusetts, USA | 14175-053 | |
| CellProfiler and CellAnalyst software packages | N/A | N/A | free open-source software |
| Centrifuge 5702 | Eppendorf AG, Hamburg, Germany | 5702000019 | |
| CO2 Incubator | HeraCell, Thermo Scientific, Waltham, Massachusetts, USA | N/A | |
| Counting chamber | BioRad Inc., Hercules, California, USA | 1450015 | |
| DeltaVision Ultra High Resolution Microscope with air table and cabinet | GE Healthcare Life Sciences, Boston, Massachusetts, USA | 29254706 | |
| Deoxyribonuclease-I (Dnase I) | Roche, Basel, Switzerland | 22098700 | |
| DMEM/F12 | Gibco, Thermo Scientific, Waltham, Massachusetts, USA | 21331–020 | |
| Dulbecco's buffer | N/A | N/A | Media kitchen, Institute of Biotechnology, University of Helsinki |
| Fetal Bovine Serum (FBS) | Gibco, Thermo Scientific, Waltham, Massachusetts, USA | 10500056 | |
| D-glucose | Sigma-Aldrich, St. Louis, Missouri, USA | G8769 | |
| Phospohate Buffer Saline (PBS) | N/A | N/A | Media kitchen, Institute of Biotechnology, University of Helsinki |
| Primocin | InvivoGen, San Diego, California, USA | ant-pm-1, ant-pm-2 | |
| poly-L-ornithine | Sigma-Aldrich, St. Louis, Missouri, USA | P4957 | |
| Triton X-100 | Sigma-Aldrich, St. Louis, Missouri, USA | 11332481001 | |
| trypsin | MP Biomedicals, Valiant Co., Yantai, Shandong, China | 2199700 |
