A Multi-Electrode Array Platform for Modeling Epilepsy Using Human Pluripotent Stem Cell-Derived Brain Assembloids
Summary
This protocol aims to stably plate dorsal-ventral fused assembloids on multi-electrode arrays for modeling epilepsy in vitro.
Abstract
Human brain organoids are three-dimensional (3D) structures derived from human pluripotent stem cells (hPSCs) that recapitulate aspects of fetal brain development. The fusion of dorsal with ventral regionally specified brain organoids in vitro generates assembloids, which have functionally integrated microcircuits with excitatory and inhibitory neurons. Due to their structural complexity and diverse population of neurons, assembloids have become a useful in vitro tool for studying aberrant network activity. Multi-electrode array (MEA) recordings serve as a method for capturing electrical field potentials, spikes, and longitudinal network dynamics from a population of neurons without compromising cell membrane integrity. However, adhering assembloids onto the electrodes for long-term recordings can be challenging due to their large size and limited contact surface area with the electrodes. Here, we demonstrate a method to plate assembloids onto MEA plates for recording electrophysiological activity over a 2-month span. Although the current protocol utilizes human cortical organoids, it can be broadly adapted to organoids differentiated to model other brain regions. This protocol establishes a robust, longitudinal, electrophysiological assay for studying the development of a neuronal network, and this platform has the potential to be used in drug screening for therapeutic development in epilepsy.
Introduction
Human pluripotent stem cell (hPSC)-derived brain organoids are spatially self-organized 3D structures mirroring the tissue architecture and developmental trajectories in vivo. They are composed of multiple cell types, including progenitors (neuroepithelial cells, radial glia, neuronal progenitors, glial progenitors), neurons (cortical-like excitatory neurons and inhibitory interneurons), and glial cells (astrocytes and oligodendrocytes)1,2. Assembloids represent the next generation of brain organoids, capable of integrating multiple brain regions and/or cell lineages within a 3D culture. They provide a useful tool for modeling connections between various brain regions resembling the in vivo counterparts, capturing interactions between neurons and astrocytes to better mimic mature and complex neural networks, and to study the assembly of neural circuits. Therefore, assembloids are becoming a widely used tool to recapitulate hallmarks of epilepsy pathophysiology, in which functional measures are necessary to interrogate aberrant neural networks that may underlie the cause of the disease3,4,5,6.
To model interactions between cortical glutamatergic neurons and GABAergic interneurons, several groups developed separate organoids resembling the dorsal and ventral brain and then fused them together into a multi-region assembloid7,8,9,10. Here, a previously described assembloid culture protocol with regionally specific neural subtypes was applied9. However, a significant barrier is the lack of reproducible functional assays to monitor neural network activity during neurodevelopment. Many functional tests of the networks in organoids yield results that have high variability among batches of differentiations and cell lines. Techniques that involve slicing or dissociating organoids alter their inherent networks by severing synaptic connectivity8.
Multi-electrode arrays (MEA) provide a large-scale view of network activity over time with high temporal resolution to characterize electrophysiological properties of organoids without disrupting culture conditions or cell membrane integrity11. Compared to patch clamp electrophysiology, MEA enables high-throughput data acquisition based on large populations of neurons rather than single cells. MEA platforms vary in their electrode density, catering to different needs in brain organoid research12. The widely used systems, as shown in this protocol, record from 8 to 64 electrodes per well13,14,15. High-density MEAs with up to 26,400 electrodes per well enable increased spatial and temporal resolution, quantification of action potential propagation speed, and combination with optogenetic stimulation14,16,17. Therefore, MEA serves as a powerful tool for modeling epilepsy in vitro and a translational paradigm for anti-seizure medication screening.
One major challenge is to stabilize the large-sized assembloids on a hydrophobic metal surface for long-term recordings. This protocol outlines a detailed methodology for plating intact assembloids on MEA plates for long-term longitudinal recording together with pharmacological assays. Unique advantages of the protocol include stable attachment of assembloids to the electrode surface without losing electrical activity, use of commercially available neurophysiological basal media to accelerate functional network maturation after plating, feasibility to conduct downstream functional assays like drug treatment, and wide application to organoids generated with other region-specific protocols.
The goal is to provide a functional assay with high temporal resolution to investigate network activity, examine disease-specific changes, and test drugs with therapeutic potential in epilepsy. Video instructions are provided for the most challenging steps of this protocol, showing the techniques for plating assembloids on MEA plates, as well as representative recordings from these cultures.
Protocol
All experimental procedures demonstrated below were conducted in accordance with the ethical guidelines of the University of Michigan Medical School Institutional Review Board and the Human Pluripotent Stem Cell Research Oversight Committee. The iPSC lines used in this protocol and the representative experiments were derived from human foreskin fibroblasts obtained from a commercial source. The details of the cell lines, reagents, and equipment used in this study are listed in the Table of Materials.
1. Derivation of fate-specific brain organoids from iPSCs
NOTE: This protocol contains information for preparing 3 wells in the microwell culture plate from 5 confluent (~85%) wells of a 6-well plate. iPSC maintenance protocol varies depending on cell lines.
- Prepare the individual cell culture media using the composition detailed in Table 1. Store protected from light at 4 °C for up to 2 weeks.
- Rinse all wells to be used in the microwell culture plate with 1 mL of DMEM/F-12 once. Add 1 mL/well mTeSR plus + 50 µM Y-27632 and spin the plate at 2000 x g for 5 min at room temperature to remove air bubbles from microwells. Store the plate in a 37 °C 5% CO2 incubator.
- Clean regions of differentiated iPSCs with a sterile P10 pipette tip under the microscope in a laminar flow clean bench. Wash off cell debris from the monolayer with 1 mL/well PBS without calcium or magnesium.
- Dissociate the monolayer into single cells by adding 1 mL/well of pre-warmed cell dissociation reagent and placing the plate in a 37 °C 5% CO2 incubator for 4-7 min (depending on characteristics and confluency of cell lines) until most cells lift off, with gentle agitation every 2-3 min.
- Add 4 mL of DMEM/F-12 to each well to dilute the cell dissociation reagent, and moderately triturate with a 10 mL serological pipette. Collect cell suspension from all wells of the same line into a 50 mL conical tube.
- Spin at 200 x g for 4 min at room temperature, aspirate the supernatant using a 10 mL serological pipette, and resuspend cells in 2 mL of mTeSR plus + 50 µM Y-27632. Take out 10 µL cell suspension, 1:1 (v/v) mix with trypan blue, and count cells with an automated cell counter.
- Plate 3 x 106 live cells per well, 3 wells per line. Add additional mTeSR plus + 50 µM Y-27632 as needed to bring the total volume to 2 mL/well. Spin the plate at 100 x g for 3 min at room temperature and put it back in the incubator.
NOTE: The microwell culture plate used here contains 300 microwells per well, so plating 3 x 106 cells results in 10,000 cells/microwell. - The next day, perform a half-volume media change by gently removing 1 mL of media and slowly adding 1 mL of mTeSR plus media (without Y-27632) with a P1000 pipette tip. Keep the tip near the media surface and aspirate as slowly as possible, as not to disturb the aggregating cells at the bottom.
- Check under an inverted microscope one day after media change, and a clear outline of each aggregate can be seen. Remove the original media from each well with P1000 pipette tips.
- Moderately spray 0.5 mL of neural induction media (NIM) with dorsomorphin (DM) and SB-4321542 (SB) into each well with sterile, cut P1000 tips. Immediately, gently pipet up aggregates suspended in media and transfer them to a sterile 10 cm suspension culture dish. Repeat until all aggregates are transferred. Do NOT triturate.
- Add additional NIM + SB/DM as needed to bring the total volume to 10 mL/dish. Put the dish on an orbital shaker in a 37 °C 5% CO2 incubator to prevent spontaneous fusion of aggregates. Mark the transfer date as Day 0 of organoid culture.
- From Day 1 to the end, follow the media recipe in Table 1 and the media change timeline9 de Figure 1. Notably, organoids are divided into two suspension culture dishes designated as "dorsal" and "ventral" on Day 4.
NOTE: Several key timepoints to keep in mind: organoid expansion begins on Day 6, when media is switched to Neural Differentiation Medium (NDM) with EGF/FGF. Neuronal differentiation is supported starting Day 25 by NDM + BDNF/NT3, and organoids are maintained in NDM without growth factors starting on Day 46.
2. Viral labeling and generation of assembloids
NOTE: Viral labeling7,9is recommended for recognizing the identity of each region-specific organoid in an assembloid and assigning electrophysiological activity from specific electrodes to assembloid regions. It is optimal if only a single organoid is plated for each MEA well. This protocol can also apply to single organoid plating.
- Calculate the volumes of virus stock and dilution media to be used: 200 µL media with virus is needed for each organoid to be labeled. Dorsal organoids are labeled with pAAV1-CAG-tdTomato, typically at a dilution ratio of 1:1000 (v/v), or approximately 2 x 1010 vg/mL. For ventral organoids, 1:300 (v/v), or 2 x 1011 vg/mL pAAV1-mDLX-GFP virus is used.
NOTE: Dilutions should be titrated to the desired effect and are affected by viral titer. - Warm up the appropriate amount of media to 37 °C. Thaw virus stock stored at -80 °C on ice. Label enough 48-well plates to accommodate all labeled organoids.
- Put a beaker containing 10% bleach in the hood. Expel any materials in contact with viruses into the beaker. Dilute the fully thawed viruses in culture media in separate 15 mL conical tubes, labeled as "dorsal" and "ventral".
- Move Day 56 organoids from culture dishes to 48-well plates. One organoid per well is recommended to prevent spontaneous fusion in static culture. Remove as much residual culture media as much as possible.
- Add 200 µL of virus-media mixture to each well, respectively. Put the plates back in the 37 °C 5% CO2 incubator. Tilt the plates on a sterile empty plate to help concentrate viral particles to the bottom of the well and optimize transduction efficiency.
NOTE: Viral particles tend to sink to the bottom of the 15 mL conical tube, so frequently inverting the tube to mix viruses with media before adding them to each well is very important to ensure uniform transduction efficiency for all organoids. - The next day (Day 57), add an additional 800 µL of culture media to each well to bring the total volume to 1 mL/well.
- Three days later (Day 60), perform a full media change on the labeled organoids. Bleach any materials in contact with viral particles.
- Three days later (Day 63), check for the fluorescence expression under an epifluorescence inverted microscope. If the signal is strong, begin generating assembloids (see step 2.9).
- Rinse all labeled organoids with 1 mL/well PBS twice to prevent cross-contamination by remaining viral particles during organoid fusion. Use a cut P1000 pipette tip to transfer one ventral organoid to each dorsal well (or vice versa) and relabel the assembloid plate accordingly. Tilt the plates as in step 2.5 for better fusion.
- Carefully perform a half-volume media change 3 days after the fusion procedure (Day 66) without interrupting the assembloids. It usually takes about a week to form stable assembloids (Day 70).
3. Placing assembloids on pre-treated MEA plates
NOTE: It takes at least 4 days to complete the surface preparation procedures of MEA plates before plating assembloids. To save time, fusing dorsal and ventral organoids (i.e., step 2.9) and MEA pre-treatment can be performed in parallel.
- Prepare reagents for surface pre-treatment and coating.
- To prepare 1% detergent/enzyme solution, dissolve 0.5 g of enzyme/detergent powder in 50 mL of deionized water. Vortex thoroughly until the powder is fully dissolved. Sterilize the solution with a sterile tube top vacuum filter in the biosafety cabinet before adding it to MEA wells. Make fresh before each use.
NOTE: The detergent/enzyme sterilizes the MEA surface, removes any residual oils from the MEA manufacturing process, and ensures that the surface is hydrophilic enough to promote interactions with the hydrophilic compound polyethyleneimine (PEI)18. - To prepare a 0.07% PEI solution19, begin with making a stock 7% PEI solution by mixing 1 mL of 50% PEI solution into a 15 mL conical tube with 6 mL of 1x borate buffer. Dilute 1 mL of stock 7% PEI solution in 99 mL of 1x borate buffer to achieve the final working concentration. For sterilization, filter through a 0.22 µm filter unit in the biosafety cabinet before use.
NOTE: PEI, a positively charged polymer, is used in the primary coating process to facilitate the attachment of a negatively charged secondary coating with Basement Membrane Matrix (BMM) and the assembloids20. Take out 1 mL of 50% PEI by pouring instead of pipetting into the conical tube, since the stock solution is very viscous. The amount needed can be more easily measured by weighing it into a conical tube, and the 50% PEI solution has a density of 1.08 g/mL. The 7% stock PEI solution can be stored in 1 mL aliquots at -20 °C for up to a month. Carry out all subsequent steps in the biosafety cabinet.
- To prepare 1% detergent/enzyme solution, dissolve 0.5 g of enzyme/detergent powder in 50 mL of deionized water. Vortex thoroughly until the powder is fully dissolved. Sterilize the solution with a sterile tube top vacuum filter in the biosafety cabinet before adding it to MEA wells. Make fresh before each use.
- In consideration of the size of each assembloid, 6-well MEA plates with 64 electrodes per well are used to accommodate one assembloid per well. Calculate the number of MEA plates needed for the experiments to ensure enough biological replicates.
- Add 1 mL of 1% detergent/enzyme solution to each well of a sterile MEA plate. Be careful not to damage electrodes with the pipette tip. If using a vacuum aspirator for washes, do not allow the tip to touch the electrode array. Incubate in a 37 °C incubator for 2 h.
- Remove detergent/enzyme and wash each well five times with 1.5 mL of sterile deionized water. Since detergent/enzyme is not biocompatible, it is important to ensure that the remaining chemicals are completely washed off.
- Fill each well with 1 mL of culture media for pre-conditioning. Put the plates back to 37 °C 5% CO2 incubator for 2 days. Exposing the MEA surface to cell culture conditions promotes cell attachment.
- Remove the culture media and cover the MEA electrodes in each well with 50 µL of 0.07% PEI solution for primary coating. Incubate at 37 °C for 1 h.
- Aspirate PEI solution completely. Wash each well with 1 mL of sterile deionized water. Perform 3 quick washes in total. Dry the plates at room temperature for 15 min in the biosafety cabinet with the lid off.
- Cover the electrode area in each well with 50 µL of 1:20 (v/v) BMM diluted in culture media for secondary coating. Put the MEA plates back in the 37 °C incubator overnight. Add sterile water to the compartments surrounding wells to ensure sufficient humidity throughout the course of coating.
- The next day, aspirate diluted BMM with a vacuum, leaving a thin layer on the surface. If a thick layer has formed, forcefully spray the electrode contact area with culture media using a P1000 pipette tip. Wash as many times as needed to remove any thick chunks.
NOTE: Residual thick BMM can interfere with electrode contact with the cells. - Collect an assembloid with a widely cut P1000 tip and place it on top of the electrodes in a well while transferring as little media as possible.
- In a laminar flow hood under a dissection scope, carefully push the assembloid with a sterile dissection needle or P20 tip to the desired location, where both dorsal and ventral organoids can be mostly covered by electrodes. Use a P20 tip to remove liquid around the assembloid. Repeat this for all assembloids or a subset, which can be done within a few minutes.
NOTE: Try NOT to touch or scratch the MEA surface with the pipette tip and/or needle. Try NOT to poke or tear the assembloid with pipette tips. Finish plating as quickly as possible to avoid the organoid completely drying out. - Incubate the assembloids at 37 °C for 10 min (without media). Transfer the plates from the incubator to a laminar flow hood and apply 2-3 small drops of 1:50 BMM diluted in culture media on top of the assembloids under a dissection scope. Incubate at 37 °C for another 30 min.
- Carefully add 3 drops of culture media close to the assembloids under a dissection scope. This keeps the organoids hydrated. Incubate at 37 °C for 1 h.
NOTE: If the assembloid moves or floats in any subsequent steps, plating should be restarted from step 3.11. - Every hour, carefully add 20-50 µL of cell culture media to each well under a dissection scope. This can be done over the course of a day. The goal is to reach at least 250 µL of total volume at the end of the day.
- The next day, check whether all assembloids are settled and stabilized under the dissection scope. Add an extra 750 µL of culture media to reach a total volume of 1 mL/well. Leave the plates undisturbed for at least 2 days.
- Carefully perform weekly media changes by taking out 500 µL of culture media and adding 700 µL of neurophysiological basal media. The additional volume of media added accounts for evaporation and can be adjusted depending on how much evaporation the MEA plate being used has. Add sterile water around the wells as needed to minimize evaporation from the MEA wells.
4. MEA recording and data analysis
- Start MEA recording about a week after switching to neurophysiological basal media (usually around Day 78).
- Wait until the recording chamber reaches its set temperature (usually 37 °C). Transfer a plate to the device and allow them to equilibrate for at least 5 min prior to recording.
- Hardware live settings
- Set the sampling frequency, i.e., the rate at which the raw voltage data is recorded. The default setting (12.5 kHz) is recommended.
- Specify analog mode (usually Neural Spikes, as in this case) to configure the hardware bandwidth and gain for particular applications. For visual monitoring of activity during recording, use Spike Detector and Burst Detector modules.
NOTE: The analog settings affect how the raw voltage data are recorded and cannot be changed after data are recorded. - (Optional) Apply a hardware digital filter to the recording data. Low pass filter options include a 2 kHz or 3 kHz Kaiser Window. Default settings can be modified after selecting the Analog settings.
- Set to referencing by median (default), which is highly recommended for neural recording, as in this case. Referencing improves the quality of low-amplitude signals by reducing the noise interference common to groups of channels.
NOTE: The parameters above can be adjusted as needed depending on the activity seen in the assembloids, but should be kept consistent for all recordings in a given experiment.
- Add plate maps and descriptions (if needed). Import the plate map from the prior recording for the same plate to ensure consistency and facilitate batch data processing. Ensure the files are named properly.
- If signals look good (minimal noise and artifact), record the raw data. Generally 2-5 min of recording is enough for a representative sample.
- Longitudinally record electrical activity once or twice a week, over 5-15 min, depending on the experimental design. Wait for at least 24 h after every media change before starting the next round of recording, as the electrical activity drops immediately after each media change.
- (Optional) If electrical activity needs to be localized and distinguished between dorsal and ventral organoids, or if the activity of each electrode needs to be analyzed separately, obtain an image of each assembloid under bright field and epifluorescence (Figure 2B) after each recording.
- When analyzing the data, apply an analysis configuration, including a spike/burst detector and statistics compiler. Use an adaptive threshold set to 5.5-6 standard deviations from baseline for spike detection. Identify bursts as an activity with ≥5 spikes in 100 ms13. Define network activity when over 25% of total active electrodes fire within 100 ms with a minimum of 50 spikes per network burst13,14.
NOTE: These parameters are modulated depending on the firing characteristics of a specific experiment. To increase sensitivity for low amplitude spikes, the adaptive threshold for spike detection can be lowered. For burst detection, various methods can be used, and the parameters can be adapted to detect bursts of spikes that differ significantly from the background activity. The network activity parameters may be modulated to a lower percentage of electrodes, especially when assembloids do not cover the entire field of electrodes. All parameters should remain consistent for the duration of an experiment to allow longitudinal comparisons. - Run the Statistics Compiler module for batch processing while replaying the recorded data. Export advanced metrics and/or spike output files for advanced analysis.
NOTE: Advanced metrics output contains .csv file with electrode, well, and group averages for mean firing, bursting, and synchrony. Spike output (.spk file) includes waveform traces for all spikes, which can be imported to other platforms for advanced analysis, e.g., spike sorting.
5. End-point pharmacological assay and plate recycling
NOTE: Drug treatment assays can be performed after assembloids are mature enough and electrical activity peaks. Recording of baseline/pre-treatment activity and post-treatment activity need to be conducted on the same day.
- Add 1-10 µL of drug solution right on top of the assembloid and gently swirl the plate to ensure a uniform mixture. Longer incubation time in drug-containing media is recommended in some cases, e.g., synapse-related drug treatment assays with glutamate/GABA receptor agonists/antagonists (Figure 2E) (see Results section for details).
NOTE: Adding a small amount of drug solution to each well is recommended to achieve a final working concentration since a large volume change will temporarily decrease electrical activity in general. It is important to ensure that the addition of a vehicle in which the drugs of interest are dissolved does not provoke significant changes in electrophysiological parameters. - Perform enough PBS washes (typically 3) and wash out drugs after the post-treatment recording. Conduct an additional recording the next day after the drug wash-out to see whether electrical activity returns to the baseline level.
- Optionally, forcefully pipet media to separate the assembloids from the MEA plate at the end of the recordings to perform further experiments, e.g., live imaging, fixation with 4% paraformaldehyde for whole-mount immunostaining, or lysis for RNA or protein extraction.
- For analysis purposes, use at least three technical and biological replicates for each condition. Express electrophysiological data as a mean of n ≥ 3 replicates.
- To recycle the plate at the end of the experiments, aspirate the remaining media after removing the attached assembloids. Wash once with sterile PBS. Fully cover the metal area with 200 µL/well of 0.25% trypsin-EDTA and put the plate back in a 37 °C incubator overnight.
- The next day, aspirate trypsin, wash twice with 70% sterile ethanol and three times with sterile deionized water.
- Air dry the plate in the biosafety cabinet for 15 min with the lid off or until it is completely dry. Seal the cleaned plate in a plastic bag and store it at room temperature until future use.
NOTE: A well-cleaned MEA plate can be reused three times without significantly losing recording quality.
Representative Results
Human dorsal-ventral assembloids were plated on a 6-well MEA plate (n = 6 per differentiation, 3 differentiations), with each well containing 64 electrodes (Figure 2A). Nine out of ten assembloids planned for longitudinal recording were firmly attached to the electrodes for over 50 days in vitro (Figure 2D). 6 out of 8 assembloids designated for pharmacological assays were successfully settled through the wash-out phase with development stage-appropriate network activity (Figure 2E). Phase contrast and fluorescence images from one representative well are shown (Figure 2B), in which the AAV-CAG-tdTomato-labeled dorsal and AAV-mDLX-GFP-labeled ventral organoids are easily distinguished7,9.
Activity of neurons on an MEA can be analyzed to assess overall cellular excitability and network activity, through a variety of parameters. Overall firing rates across the culture provide information on cellular and network excitability, while the frequency of synchronized bursts across multiple electrodes indicates the range of synaptically active networks of neurons. Multiple repeated spikes occurring across spatially distinct electrodes over a short period of time (network bursting) can serve as a biological correlate to seizure-like activity, although assembloids on MEAs remain limited compared to a whole organism that can have bona fide seizures. Figure 2C illustrates the network bursting patterns of assembloids cultured in commercially available neurophysiological basal media on Days 84 and 91, with increased bursting duration at the later timepoint likely due to functional maturation of the neurons. In this case, cellular and network activity gradually rises from the day when assembloids are plated, usually peaking on Day 92, and almost fading away on Day 125 (Figure 2D). The increased maturation at the later stage likely contributes to the extended synchronized burst duration at the later recording time point.
Bicuculline, a GABAA receptor antagonist, enhances the synchronized network activity of the assembloids, as shown by significantly higher firing rates and (network) burst frequencies after adding the drug on Day 108 (Figure 2E). This is similar to what has been previously demonstrated in single-cell electrophysiology recordings21. A power analysis for the pharmacological assay was conducted using G*Power3 base on an effect size of 0.94, and an alpha of 0.05. A total sample of 15 assembloids with three equal-sized groups of n = 5 was required to achieve a power of 0.8 (Figure 2E). A vehicle control is included to verify that the response is a true signaling event and not merely a change in signal intensity that may arise from the addition of a small amount of media (Figure 2E).
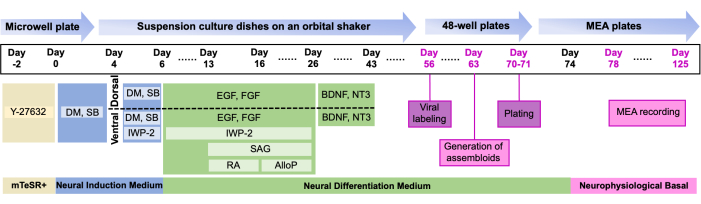
Figure 1: Overview of the study. Protocol for the generation and plating of dual labeled assembloids on MEA timeline of the differentiation protocol detailing critical stages and corresponding small-molecule supplements (for additional information on the composition, see Table 1 and Table of Materials). Please click here to view a larger version of this figure.
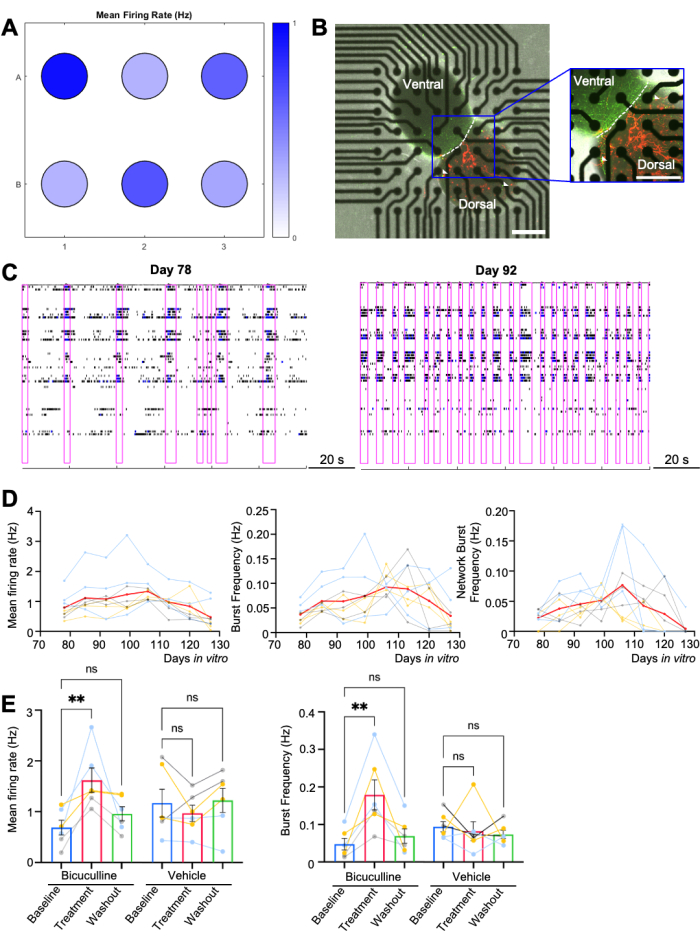
Figure 2: MEA recording of assembloids. (A) Heat map of mean firing activity from a single MEA plate (n = 6 wells) at Day 82, i.e., post-plating day 10. All wells represent cultures of the same condition and genotype. (B) Representative phase-contrast and confocal images of the assembloid, with dorsal assembloid labeled with red and ventral with green. White arrowheads indicate migrated GABAergic interneurons from the ventral to the dorsal side. Scale bar = 500 µm. (C) Representative raster plots over 120 s of recording time from a single well. Network bursts are highlighted with a purple box. Two separate timepoints, Day 78 (1 week after plating, starting day of recording) and Day 92 (3 weeks after plating, also when the activity peaks), are demonstrated. (D) Quantification of mean firing rates, burst frequency, and network bursting frequency from day 78 to day 127 (n = 3 differentiations, 3 assembloids/ differentiation). Each curve indicates one individual assembloid, compiled from 3 differentiations (color-coded). The red curve demonstrates the mean value. (E) Quantification of bicuculline assay results (n = 6 assembloids/ condition). The bar graph was displayed as mean ± SEM. Each dot represents one assembloid, compiled from 3 batches of differentiation (color-coded). A nonparametric Friedman test was performed. **p < 0.01; ns, not significant. MEA, multi-electrode arrays. Heat map and raster plots are generated using the analytic software Neural Metric Tool. Please click here to view a larger version of this figure.
Table 1: Components of media at various differentiation stages. Please click here to download this Table.
Discussion
MEA-based methods for electrophysiological recordings of network activity in iPSC-derived assembloids have been used for in vitro modeling of epilepsy22,23. This proposed platform integrating excitatory and inhibitory synaptic connections has the potential to address mechanisms of neuronal hyperexcitability and the role of cortical interneurons in the process of epileptogenesis. Additionally, this platform allows for the stabilization of assembloids and collection of longitudinal electrophysiological data for about 50 days in vitro, even in the presence of pharmacological perturbations, thereby providing an approach for testing compounds with potential therapeutic targets.
Stabilizing mature, relatively large assembloids on a hydrophobic surface for a long period in vitro has been technically challenging. To overcome this obstacle, the twice-coating strategy is applied with conversely charged chemicals to facilitate short- and long-term adherence of organoids to the electrode surface. This protocol optimizes the previously used method24 by switching the plate-coating reagents from poly-L-ornithine (PLO)/laminin to PEI/BMM, achieving solid attachment with high success rates and low toxicity. Furthermore, this stabilized long-term culture system has the potential to yield high-throughput data. For example, pre-labeling with dual viruses can complement spike sorting by individual electrodes to localize and compare activity from different regions. The proposed platform also provides a stable tool for recording local field potentials, which is highly relevant to interneuron function without disrupting the intact structure of assembloids25. The major limitation of this protocol is that assembloids are flattened after plating, which raises concerns about whether the initial 3D structures are preserved or not. Growing organoids around the electrodes using more advanced MEA platforms that envelope an organoid with electrodes26 could potentially overcome the limitation.
One of the main obstacles of MEA-based methods for electrophysiological recording of organoids is the variability and reproducibility of experimental findings across differentiation protocols and even batches of differentiation. Previous studies show that the network activity of neurons measured by MEA is dependent upon the neuronal maturation status of both dorsal and ventral organoids27. Recording from assembloids at a later timepoint with relatively mature neurons and circuits is one way to improve consistency. Furthermore, the choice of multi-well MEA systems generating high throughput data will allow statistical comparisons in light of experimental variability. If the electrophysiological activity of a culture is inconsistent with what is expected at a given time point, collecting the intact culture from the functional platform for quality control measures, including immunocytochemical and transcriptomic analysis, will be important and helpful.
Paramount to the proposed platform is to functionally integrate regionally specified excitatory cortical neurons on the dorsal side and inhibitory interneurons on the ventral side. Protocols for the generation of brain region-specific organoids found in the literature differ extensively in relation to media conditions, the timing of cultures, yield, and maturation profiles, among other factors28. While this protocol is cortex- and ganglionic eminence-specific in its ability to generate excitatory neurons and GABAergic interneurons, well-established differentiation techniques can be used to generate iPSC-derived neurons displaying other identities, such as motor neurons29 and Purkinje cells30. Thus, the proposed platform has the potential to model neural circuits between the cerebral cortex and the spinal cord/ cerebellum. After some optimization related to the timing of plating and visualization of cell types with viral labeling, this method can be translated to other disease models aside from epilepsy, like Amyotrophic Lateral Sclerosis (ALS)17.
Divulgations
The authors have nothing to disclose.
Acknowledgements
This manuscript was supported by R01NS127829 NIH/NINDS (LTD). Figure 1 has been generated using biorender.com.
Materials
| 10 cm Corning Non TC-treated culture dishes | Corning | 08-772-32 | For suspension culture on the shaker |
| 100 mL Beaker | Fisher Scientific | FB100100 | |
| 100% Ethanol | Fisher Scientific | BP28184-4L | |
| 2-Mercaptoethanol (β-ME) | Thermo Fisher | 21985023 | Working concentration 100 μM |
| 48-well cell culture plate | Fisher Scientific | 50-202-140 | |
| 6-well cell culture plate | Fisher Scientific | 07-200-83 | |
| Aggrewell 800 | Fisher Scientific | 501974754 | |
| Allegra X-14R Refrigerated Centrifuge | Beckman Coulter | BE-AX14R | |
| Allopregnanolone | Cayman | 16930 | Suspended 5mg into DMSO to get 1 mM stock solution. Aliquot and freeze at −80 °C. Dilute at 1:10,000 for use. Working concentration 100 nM. |
| Automated cell counter | Thermo Fisher | AMQAX2000 | |
| Axion CytoView MEA 6-well plates | Axion Biosystems | M384-tMEA-6B | |
| Axion Maestro MEA platform | Axion Biosystems | Maestro | With temp environmental control |
| B-27 supplement (regular, with Vitamin A) | Thermo Fisher | 21985023 | |
| B-27 supplement (without Vitamin A) | Thermo Fisher | 12587010 | |
| Basement membrane matrix- Geltrex | Thermo Fisher | A1569601 | |
| Bead bath | Fisher Scientific | 10-876-001 | Isotemp |
| Benchtop inverted microscope | Olympus | CKX53 | Kept in laminar flow clean bench |
| Bicuculline | Sigma-Aldrich | 14340 | Working concentration 10 μM |
| Bleach | CLOROX | 67619-26 | |
| Borate buffer 20x | Thermo Fisher | 28341 | Working concentration at 1x |
| BrainPhys media | StemCell Technologies | 5790 | |
| Cell dissociation reagent (StemPro Accutase) | Thermo Fisher | A1110501 | |
| Celltron orbital shaker | HT-Infors | I69222 | |
| Detergent/enzyme (Terg-A-Zyme) | Sigma-Aldrich | Z273287 | Working concentration 1% m/v |
| DMEM/F12 + HEPES/L-Glutamine | Thermo Fisher | 113300 | |
| DMSO | Sigma-Aldrich | 67685 | |
| Dorsomorphin | Sigma-Aldrich | P5499 | Dissolve 5mg into DMSO to get 10 mM stock solution. Aliquot and freeze at −20 °C. Dilute at 1:2000 for use. (working concentration 5 μM) |
| D-PBS w/o calcium or magnesium | Thermo Fisher | 14190144 | |
| Glial cell line-derived neurotrophic (GDNF) | Peprotech | 450-10 | Dissolve 100 μg in 1mL of PBS to 100 μg/mL. Aliquot and freeze at −20 °C. Dilute at 1:5000 for use. (working concentration 20 ng/mL). |
| GlutaMAX supplement | Thermo Fisher | 35050061 | |
| Hemacytometer | Election Microscopy Sciences | 63510-20 | |
| HEPES | Thermo Fisher | 15630080 | |
| Heraguard ECO Clean Bench | Thermo Fisher | 51029692 | |
| Humidity controlled cell culture incubator | Thermo Fisher | 370 | set to 37 °C, 5% CO2 |
| IWP-2 | Selleckchem | S7085 | Aliquot and freeze at −80 °C. It will precipitate if thawed at room temp. Frozen aliquots should be placed directly into 37 °C before use. |
| Knockout serum replacement (KOSR) | Thermo Fisher | 10828010 | |
| mTeSRplus (medium + supplements) | StemCell Technologies | 100-0276 | cGMP, stabilized feeder-free medium for human iPSC cells |
| N2 supplement | Thermo Fisher | 17502048 | |
| Neurobasal A | Thermo Fisher | 21103049 | |
| Non-essential amino acids (NEAA) | Thermo Fisher | 11140050 | |
| NT3 | Peprotech | 450-03 | Dissolve 100 μg in 1mL of PBS to 100 μg/mL. Aliquot and freeze at −20 °C. Dilute at 1:5000 for use. (working concentration 20 ng/mL). |
| NuFF Human neonatal foreskin fibroblasts | MTI-GlobalStem | GSC-3002 | |
| Parafilm | PARAFILM | P7793 | |
| Penicilin/Streptomycin | Thermo Fisher | 15140122 | |
| Pipette (P10, P200, P1000) | Eppendorf | EP4926000034 | Autoclaved cut P1000 tips for organoid collection |
| Poly (Ethyleneimine) (PEI) | Sigma-Aldrich | P3143 | Dilute stock in sterile borate buffer. Working concentration 0.07%. See details in manuscript. |
| Recombinant human epidermal growth factor (EGF) | R&D Systems | 236-EG-200 | Suspended in PBS. Aliquot and freeze at -20 °C. |
| Recombinant Human fibroblast growth factor (FGF)-basic | Peprotech | 100-18B | Suspended in PBS. Aliquot and freeze at -20 °C. |
| Recombinant human-brain-derived neurotrophic factor (BDNF) | Peprotech | 450-02 | Centrifuge briefly before reconstitution. Dissolve 100 μg in 1 mL of PBS to 100 μg/mL. Aliquot and freeze at −20 °C. Dilute at 1:5000 for use. (working concentration 20 ng/mL). |
| Retinoic acid (RA) | Sigma-Aldrich | R2625 | Dissolve 100 mg into 3.3 mL of DMSO to get 100 mM stock solution. Aliquot the stock 100 μL/tube and freeze at −80 °C. Take 200 μL of 100 mM stock and dilute 10x (add 1.8 mL of DMSO) to make 10 mM stock. Aliquot 50 μL/tube and store at −80 °C. Dilute at 1:100,000 for use. (working concentration 100 nM). |
| ROCK inhibitor Y-27632 | Tocris | 1254 | 1:200 from 10 mM stock |
| SAG (smoothened agonist) | Selleckchem | S7779 | Aliquot and freeze at −80 °C. Stock concentration 1mM. Use at 1:10,000 dilution (working concentration 100 nM). |
| SB-431542 | Tocris | 1614 | Dissolve 5mg into 1.3 mL of DMSO to get 10 mM stock solution. Aliquot and freeze at −80 °C. Dilute at 1:1000 for use. (working concentration 10 μM) |
| Serological pipette filler | Fisher Scientific | 14-387-166 | |
| Steriflip vacuum tube top filter | Sigma-Aldrich | SE1M179M6 | |
| Sterile cell culture hoods | Baker Company | SG-600 | |
| Trypan blue solution (0.4%) | Thermo Fisher | 15250061 | |
| Trypsin-EDTA (0.25%) | Thermo Fisher | 25200056 | |
| Zoom stereomicroscope | Olympus | SZ61/SZ51 | Kept in laminar flow clean bench |
References
- Lancaster, M. A., Knoblich, J. A. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science. 345 (6194), 1247125 (2014).
- Tanaka, Y., et al. Synthetic analyses of single-cell transcriptomes from multiple brain organoids and fetal brain. Cell Rep. 30 (6), 1682-1689.e3 (2020).
- Meng, X., et al. Assembloid CRISPR screens reveal impact of disease genes in human neurodevelopment. Nature. 622 (7982), 359-366 (2023).
- Samarasinghe, R. A., et al. Identification of neural oscillations and epileptiform changes in human brain organoids. Nat Neurosci. 24 (10), 1488-1500 (2021).
- Saleem, A., et al. Modelling hyperexcitability in human cerebral cortical organoids: Oxygen/glucose deprivation most effective stimulant. Heliyon. 9 (4), e14999 (2023).
- Saberi, A., et al. In-vitro engineered human cerebral tissues mimic pathological circuit disturbances in 3D. Commun Biol. 5 (1), 1-9 (2022).
- Bagley, J. A., et al. Fused dorsal-ventral cerebral organoids model complex interactions between diverse brain regions. Nat Methods. 14 (7), 743-751 (2017).
- Birey, F., et al. Assembly of functionally integrated human forebrain spheroids. Nature. 545 (7652), 54-59 (2017).
- Sloan, S. A., et al. Generation and assembly of human brain region-specific three-dimensional cultures. Nat Protoc. 13 (9), 2062-2085 (2018).
- Xiang, Y., et al. Fusion of regionally-specified hPSC-derived organoids models human brain development and interneuron migration. Cell Stem Cell. 21 (3), 383-398.e7 (2017).
- VanDersarl, J. J., Renaud, P. Biomimetic surface patterning for long-term transmembrane access. Sci Rep. 6 (1), 32485 (2016).
- Sandoval, S. O., et al. Rigor and reproducibility in human brain organoid research: Where we are and where we need to go. Stem Cell Rep. 19 (6), 796-816 (2024).
- Hartmann, J., et al. Molecular and functional characterization of different brainsphere models for use in neurotoxicity testing on microelectrode arrays. Cells. 12 (9), 1270 (2023).
- Chen, Y., et al. Generation of advanced cerebellar organoids for neurogenesis and neuronal network development. Hum Mol Genet. 32 (18), 2832-2841 (2023).
- Fair, S. R., et al. Electrophysiological maturation of cerebral organoids correlates with dynamic morphological and cellular development. Stem Cell Rep. 15 (4), 855-868 (2020).
- Muzzi, L., et al. Human-derived cortical neurospheroids coupled to passive, high-density and 3D MEAs: A valid platform for functional tests. Bioeng. (Basel). 10 (4), 449 (2023).
- Hruska-Plochan, M., et al. A model of human neural networks reveals NPTX2 pathology in ALS and FTLD. Nature. 626 (8001), 1073-1083 (2024).
- Reumann, D., et al. In vitro modeling of the human dopaminergic system using spatially arranged ventral midbrain-striatum-cortex assembloids. Nat Methods. 20 (12), 2034-2047 (2023).
- Fenton, T. A., et al. Hyperexcitability and translational phenotypes in a preclinical model of SYNGAP1 mutations. bioRxiv. , (2023).
- Huang, Q., et al. Shell microelectrode arrays (MEAs) for brain organoids. Sci Adv. 8 (33), eabq5031 (2022).
- Dong, X., et al. Human cerebral organoids establish subcortical projections in the mouse brain after transplantation. Mol Psychiatry. 26 (7), 2964-2976 (2021).
- Patton, M. H., et al. Synaptic plasticity in human thalamocortical assembloids. bioRxiv. , (2024).
- Osaki, T., et al. Complex activity and short-term plasticity of human cerebral organoids reciprocally connected with axons. Nat Commun. 15 (1), 2945 (2024).
- Trujillo, C. A., et al. Complex oscillatory waves emerging from cortical organoids model early human brain network development. Cell Stem Cell. 25 (4), 558-569.e7 (2019).
- Schröter, M., et al. Functional imaging of brain organoids using high-density microelectrode arrays. MRS Bull. 47 (6), 530-544 (2022).
- Nieto-Estevez, V., et al. Dual effects of ARX poly-alanine mutations in human cortical and interneuron development. bioRxiv. , (2024).
- Ciarpella, F., et al. Murine cerebral organoids develop network of functional neurons and hippocampal brain region identity. iScience. 24 (12), 103438 (2021).
- Zhang, Z., et al. Development and application of brain region-specific organoids for investigating psychiatric disorders. Biol Psychiatry. 93 (7), 594-605 (2023).
- Andersen, J., et al. Generation of functional human 3D cortico-motor assembloids. Cell. 183 (7), 1913-1929.e26 (2020).
- Atamian, A., et al. Human cerebellar organoids with functional Purkinje cells. Cell Stem Cell. 31 (1), 39-51.e6 (2024).
Tags

.