Mycobacterium abscessus (Mab) is an opportunistic pathogen responsible for pulmonary infections, especially in people with cystic fibrosis and other lung disorders. Infections caused by Mab are infamously challenging to treat due to exquisite intrinsic drug resistance, comparable to multidrug-resistant tuberculosis1. The available drugs are largely ineffective due to the highly impermeable mycobacterial cell envelope and a genome that encodes several enzymes that deactivate antibiotics2. Thus, treatment includes the combination of several drugs taking months to years. This challenging multi-drug regimen combined with low patient compliance results in an average cure rate of 30% to 50%3. Furthermore, the prevalence of pulmonary infections caused by non-tuberculous mycobacteria has increased over the past few decades, including the ones caused by Mab1,4. Consequently, the scientific community is racing to develop new compounds to treat Mab infections.
One of the strategies pursued with this aim is drug repurposing – the process of identifying new therapeutic opportunities for existing drugs. This circumvents the biggest challenge accompanying a new drug's discovery and development pipeline – time5. This simple concept takes advantage of the already established pharmacokinetic and safety profiles of several drugs to reduce developmental costs and shorten the timescale that it takes to get a drug from bench to bedside6. Thus, libraries that combine hundreds to thousands of such compounds have been compiled, allowing researchers to quickly test the possibility of repurposing drugs against their pathogen of interest.
Most studies on drug development against Mab are based around a gold-standard yet traditional assay that evaluates a compound's in vitro activity against mycobacteria – colony-forming units counting7. Despite its accuracy, this procedure is extremely time-consuming, quickly becoming non-feasible when someone aims to test libraries containing thousands of compounds. To this end, high-throughput screenings (HTS) have been integrated into drug development – robust assays that take advantage of robotics and liquid handling devices, allowing thousands of compounds to be quickly screened in parallel8. This is typically done by testing a single concentration initially, using microtiter plates of 96-, 384-, 1536-, or 3456- well formats, acting as a starting point to identify hits and further optimize them down the pipeline to be clinically used.
Reporter-based assays provide a significant advantage for the robustness of HTS due to their simplicity and sensitivity compared to other dye and absorbance-based assays7,9. However, to our knowledge, only a few studies have optimized a high-throughput screening against Mab9.
Recently, our lab has developed double reporter strains capable of concomitantly emitting luminescence and fluorescence10. Mab operon_mScarlet is one of those strains. It is auto-luminescent due to the expression of the LuxABCDE operon, which includes a bacterial luciferase (by expression of the luxAB genes) and a long-chain aldehyde substrate (by expression of the luxCDE genes). On the other hand, the fluorescent readout is obtained through the expression of a recently developed red fluorescent protein, mScarlet, which outperforms the more typically used eGFP and mCherry proteins, providing a more potent signal11. Using this strain enables us to assess the bacterial viability in liquid culture by measuring the luminescent signal in a microplate reader without adding reagents or performing additional steps. In terms of detection, intrinsic fluorescence allows for microscope visualization in live or fixed cells without using dyes or antibodies. Having one single strain with both readouts provides researchers with a significant advantage when using it in HTS assays – the reduced variability between assays with different readouts, as there is no need to exchange strains based on the nature of the assay.
Thus, this work developed a high-throughput assay against Mab using an in house engineered double reporter strain (Figure 1). This allowed for a quick assessment of the in vitro activity 1280 drugs intended for repurposing using a commercial library (see Table of Materials). Firstly, the activities were assessed in broth culture assay using luminescence, and secondly, by using Mab-infected macrophages taking advantage of the fluorescent signal, better emulating the infection process seen in vivo12.
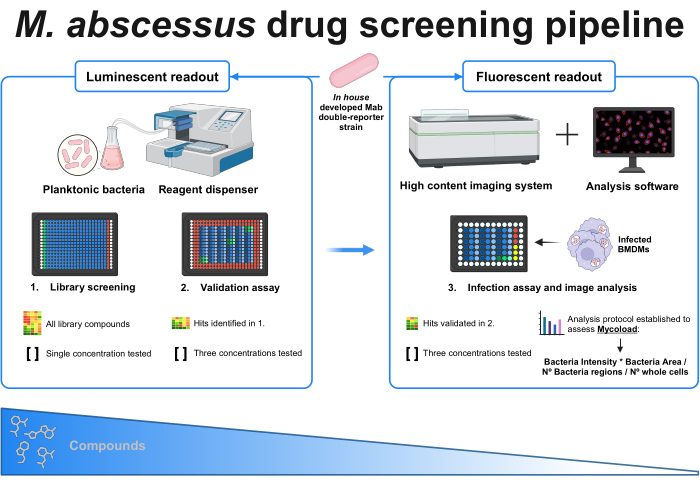
Figure 1: Graphical abstract of the established protocol. The key player in this screening is an in house developed double reporter mycobacterial strain, which is used throughout all experiments. Firstly, using a reagent dispenser and planktonic bacteria, an initial screening is performed by testing the compounds at a single concentration. The identified hits carry on to the validation assay, where three different concentrations are tested. Both experiments are done using the luminescent readout. The validated hits carry on to the infection assay, where three different concentrations are also tested, and bone marrow-derived macrophages are infected at MOI 1. A high-content imaging system acquires the bacterial fluorescent signal, and analysis software is used to assess the intracellular load through the Mycoload formula. The number of compounds reduces as assay complexity increases. Created with BioRender.com. Please click here to view a larger version of this figure.
All animal procedures were approved by the Local Animal Ethics Committee of i3S and licensed by the Portuguese Authority General Directorate for Food and Veterinary (DGAV), following the European Council Directive (2010/63/EU) and the Portuguese law (DL 113/2013) for animal welfare on experimentation.
1. Z' factor assessment
NOTE: To assess the performance of an HTS assay, several parameters can be determined. The most used parameter is the Z prime factor (Z' factor). This metric measures the statistical effect size of a given experiment, indicating how well separated positive and negative controls are. If a Z' factor ranges between 0.5 and 1, the separation between both populations is excellent, and these conditions can be used in the HTS assay. If Z' < 0.5, the separation is only marginal, and it is not recommended to perform the screening13. As such, an experiment using half a plate with non-treated bacteria and the other half with a positive control to calculate the Z' factor was designed (Supplementary Figure 1).
- At 2-3 days before the assay, prepare pre-cultures of Mab by inoculating bacteria in a liquid growth medium (approximately 1 x 107 CFU/mL) in an Erlenmeyer flask. Leave the bacteria to grow at 37 °C in an incubator with agitation (90 rpm).
CAUTION: A biosafety cabinet is required when working with Mab cultures. - Follow the Mab preculture's growth by optical density measurement at 600 nm using a spectrophotometer. When the culture reaches the exponential growth phase, carry on to step 1.3.
- Using the exponential pre-culture, prepare a bacterial suspension of approximately 5 x 105 CFUs/mL (correlated with optical density values)14 in liquid growth medium.
- Prepare a solution of positive control of bacterial growth inhibition in a liquid growth medium. For this assay, the positive control was clarithromycin (10 mg/mL in DMSO) at 4 µg/mL. Clarithromycin must be prepared 2x the desired final concentration.
- Using a microplate reagent dispenser equipped with a small cassette, dispense 15 µL of liquid growth medium to the blank and non-treated columns of a black polypropylene 384-well plate. For the blank columns, dispense an additional 15 µL of liquid growth medium.
NOTE: The blank columns prevent evaporation of the inner wells at 37 °C. - With the microplate reagent dispenser equipped with a small cassette, dispense 15 µL of the positive control solution prepared in step 1.4 to the positive control columns.
- With a microplate reagent dispenser equipped with a large cassette, dispense 15 µL of the bacterial suspension prepared in step 1.3 to all columns except blanks, diluting everything 1:2. The final volume of all 384 wells is 30 µL.
NOTE: The microplate reagent dispenser can be equipped with a large or small cassette, differing in tubing size (0.5 and 0.22 mm, respectively). It is preferable to use a bigger cassette to prevent bacterial aggregation in the tubing system, even though resulting in more liquid wastage. Nonetheless, the same cassette can be used throughout the whole assay. - Seal each plate with a plate sealer that allows continuous gas exchange. Incubate at 37 °C in an incubator without agitation inside a humid chamber for 48 h.
- After 48 h, read the luminescence emitted by the bacteria in each plate using a plate reader with an integration time of 1 s.
- To determine the Z' factor, use the values obtained in step 1.9 and calculate the average and standard deviation of the positive (AVGP and SDP) and negative (AVGN and SDN) controls. Afterward, apply the following formula:
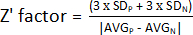
NOTE: 3 x SD is a threshold commonly used; however, it can be adjusted to better suit the screening – either by increasing or decreasing the factor by which to multiply the standard deviation.
2. Library screening for hits
NOTE: The first screenings were performed against bacteria growing in liquid culture. The setup was designed for 384-well plates with 30 µL of final volume. Each plate contained blank wells (liquid growth medium only), negative controls (non-treated bacteria), and solvent control (bacteria plus compound's solvent – in this case, DMSO; Supplementary Figure 2). The compounds or solvent were added to the plate at 2x the desired final concentration, and bacteria were added 1:1 (diluting everything to half).
- Prepare bacterial suspension as described in steps 1.1 to 1.3.
CAUTION: A biosafety cabinet is required when working with Mab cultures. - Prepare a solution of solvent in a liquid growth medium. The amount of solvent must be 2x the desired final concentration.
- Using a microplate reagent dispenser equipped with a small cassette, dispense 15 µL of liquid growth medium to every column of black polypropylene 384-well plates, except for the solvent control column. For blank columns, dispense an additional 15 µL of liquid growth medium.
- With the microplate reagent dispenser equipped with a small cassette, dispense 15 µL of the solvent solution previously prepared to the solvent control column.
- Using a liquid handler, transfer 200 nL of the compounds from the stock 96-well plates (1 mM in DMSO) to the 384-well plates, except for the blanks, solvent control, and non-treated columns. The compounds are now at a concentration of 13.3 µM.
- With a microplate reagent dispenser equipped with a large cassette, dispense 15 µL of the bacterial suspension prepared in step 2.1 to all columns except blanks, diluting everything 1:2. The final volume of all 384 wells is 30 µL, and each compound is at 6.66 µM.
- Seal each plate with a plate sealer that allows continuous gas exchange.
NOTE: The assay can be done with regular lids; however, there is a higher possibility of stronger edge effects. - Incubate at 37 °C in an incubator without agitation inside a humid chamber for 48 h.
- After 48 h, read the luminescence emitted by the bacteria in each plate using a plate reader with an integration time of 1 s.
3. Hit screening data analysis
NOTE: To identify hits with the acquired luminescence data, the values must be normalized to the solvent in which the compounds are dissolved and, if it occurs, to the edge effect verified on the 384-microtiter plates.
- For the solvent normalization of the data obtained in step 2.9, divide each well's relative light units (RLU) by the average RLU of the solvent control wells (in the corresponding plate). In this assay, a strong edge effect was verified towards the middle of the screening plates; thus, it was decided to normalize all data for it. To achieve this, the process was done in a two-step manner, as described below.
- Create an edge effect mask by dividing the values obtained in step 3.1 for the corresponding positions, excluding hits, across all plates (plate 1 – P1, plate 2 – P2, plate 3 – P3, plate 4 – P4). For example, for A1 and A2 positions, Mask_A1 = Average (P1_A1; P2_A1; P3_A1; P4_A1); Mask_A2 = Average (P1_A2; P2_A2; P4_A2) [P3_A2 was excluded because it was a hit].
- Divide the values obtained in step 3.1. by its edge effect mask obtained in step 3.1.1. For example,
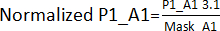 . Afterward, calculate each plate's AVG and SD to establish a threshold (AVG ± 3SD) for each plate. Any compound outside this threshold is deemed a hit and carries on to the hit validation assay.
. Afterward, calculate each plate's AVG and SD to establish a threshold (AVG ± 3SD) for each plate. Any compound outside this threshold is deemed a hit and carries on to the hit validation assay.
4. Hit validation assay
NOTE: After identifying hits, these need to be validated using different concentrations. The validation assay uses a similar setup with the same controls, but each hit will be subjected to 1:2 serial dilutions. For this assay, three concentrations were used – 13.3 µM, 6.66 µM, and 3.3 µM. Solvent controls will contain the same solvent % as each compound well (Supplementary Figure 3).
- Prepare bacterial suspension as described in steps 1.1 to 1.3.
CAUTION: A biosafety cabinet is required when working with Mab cultures. - Using a microplate reagent dispenser equipped with a small cassette, dispense 15 µL of liquid growth medium to every column of a black polypropylene 384-well plate. For blank wells, dispense an additional 15 µL of liquid growth medium to the first and last column.
- With the microplate reagent dispenser, dispense an additional 15 µL of liquid growth medium to the columns containing the highest [µM] of each compound.
- From the stock 96-well plates, manually pipette 0.8 µL of each hit (1 mM in DMSO) into their respective wells from step 4.3, in duplicate. The compounds are now at a concentration of 26.6 µM.
- Manually pipette 0.8 µL of solvent into the control wells in duplicate (same as step 4.4).
- Using a multi-channel micropipette, perform two 1:2 serial dilutions for each compound and solvent control. Discard the exceeding 15 µL after the last serial dilution. All wells now have 15 µL, and the concentrations of the compounds are 26.6 µM, 13.3 µM and 6.66 µM.
- Using a microplate reagent dispenser with a large cassette, dispense 15 µL of the bacterial suspension prepared in 4.1 to all columns (except blank wells), diluting the compounds 1:2. All wells have a final volume of 30 µL, and each compound is at 13.3 µM, 6.66 µM, and 3.3 µM.
- Seal the plate with a plate sealer that allows continuous gas exchange. Incubate at 37 °C in an incubator without agitation inside a humid chamber for 48 h.
- After 48 h, read the luminescence emitted by the bacteria in each plate with an integration time of 1 s using a plate reader.
5. Infection assay
NOTE: Mab is an intracellular facultative pathogen, so the hits' antimicrobial activity and host toxicity must be determined in infection assays. For that, bone marrow-derived macrophages (BMM) were infected with Mab and treated with the hits validated in step 4 at different concentrations. Each assay included blank (only water), negative controls (non-treated infected macrophages), positive controls (infected macrophages treated with an effective antibiotic), solvent controls (in this case, DMSO), and the compounds to be tested in three concentrations – 13.3 µM, 6.66 µM, and 3.3 µM (Supplementary Figure 4).
- Derive macrophages from the murine bone marrow of wild-type adult mice (BALB/c or C57BL/6), as described previously15. Plate the cells on a 96-well, black, optically clear flat-bottom plate at 2 x 105 cells/mL, 200 µL per well. After 10 days of differentiation at 37 °C, 7% CO215, the cells are ready to infect and proceed with treatment.
NOTE: The protocol for obtaining bone marrow-derived macrophages is well-established15; however, this screening protocol can be adapted to other cells, such as RAW 264.7, THP-1, and PBMC-derived macrophages. - Prepare bacterial suspension as described in steps 1.1 to 1.3, adjusting the bacterial suspension in cell culture medium for an infection with a multiplicity of infection (MOI) of 1.
CAUTION: A biosafety cabinet is required when working with Mab cultures. - Carefully aspirate the supplemented cell culture medium of the wells using a vacuum pump attached to a glass pipette. This step needs to be performed slowly and with ease to prevent cell detachment.
- Manually pipette 75 µL of the bacterial suspension prepared in step 5.2 to each well. Incubate the plate at 37 °C with 7% CO2 for 4 h in a cell incubator.
- Prepare the compounds for cell treatment. Using a microplate reagent dispenser equipped with a small cassette, dispense to polypropylene, round bottom, 96-well plates, 110 µL of supplemented cell culture medium to columns where compounds or solvent control will be (Supplementary Figure 4).
- Using a microplate reagent dispenser, dispense an additional 104 µL of supplemented cell culture medium to the columns where the highest concentration of compound or solvent will be.
- Manually pipette 5.9 µL of the compounds (1 mM in DMSO) or solvent into their designated wells (columns in step 5.6). The compounds are now at a concentration of 26.6 µM.
- Using a multi-channel micropipette, perform two serial dilutions 1:2 for each compound and solvent. Discard the exceeding 110 µL after the last serial dilution. All wells now have 110 µL, and the compounds are at a concentration of 26.6 µM, 13.3 µM, and 6.66 µM.
- Using a microplate reagent dispenser, dispense 110 µL of supplemented cell culture medium to all columns (excluding blank wells). All wells now have 220 µL, and the compounds are at a concentration of 13.3 µM, 6.66 µM, and 3.3 µM.
- Prepare a solution of clarithromycin (positive control) at 2 µg/mL in a supplemented cell culture medium.
- At 4 h after step 5.4, using a plate washer, wash 3x with 200 µL of infection washing solution the infected 96-well plate. Keep the aspiration and dispense velocities to the slowest possible to prevent any cell detachment.
- After the final aspiration, transfer 200 µL of the compounds prepared in steps 5.5-5.9 to the 96-well plate containing the infected macrophages.
- Transfer 200 µL of supplemented cell culture medium to the non-treated wells. Transfer 200 µL of the clarithromycin solution prepared in step 5.10 to its respective wells.
- Incubate the plate in a cell incubator at 37 °C, with 7% CO2, for 48 h.
- After 48 h, wash the infected 96-well plate using a plate washer with the compound's washing solution (3x with 200 µL per well).
- Using a multi-channel micropipette, dispense 200 µL of the fixation solution to all wells and let it act for 10 minutes.
- After fixation, using a plate washer, wash the cells with the compounds' washing solution (3x with 200 µL per well).
NOTE: The protocol can be paused here. The cells can be stored in the compounds' washing solution at 4 °C for several days or weeks if contamination and cell dryness are prevented. The compounds' washing solution must be aspirated before step 5.18. - Using a multi-channel micropipette, dispense 200 µL of the permeabilizing solution to all wells and let it act for 15 min. Using a plate washer, aspirate the permeabilizing solution.
- Using a multi-channel micropipette, manually dispense 100 µL of the staining solution and incubate the plates at room temperature for 30 min.
- Using a plate washer, wash the plates with the compounds' washing solution (3x with 200 µL per well). In the end, leave the stained cells in the compounds' washing solution.
- Screen the plates in a high-content imaging system with the following settings:
Objective: 20x Air/0.4NA (one well of a 96-microtiter plate, 57 fields of view (FOV)
Mode: Confocal
Laser 405 nm for DAPI (nuclei)
Laser 561 nm for mScarlet (bacteria)
Laser 640 nm for HCS Cell Mask Deep Red (cytoplasm)
No z-stack
NOTE: The cells can be imaged with a widefield imaging system, decreasing the definition. - Set up an equipment handling robot for image acquisition overnight. This robotic arm exchanges the plates in the high-content imaging system without human intervention.
NOTE: This is not mandatory; plates can be exchanged manually.
6. Image analysis
NOTE: The image analysis is performed with the analysis software, which is the same as the high-content imaging system used to acquire the images. An analysis pipeline must be created using sample images (Supplementary Figure 5). Afterward, it will be applied to the entire well (57 FOV) and dataset (all plates). The analysis software follows a logical sequence of steps, starting with the segmentation of the different regions of interest (nuclei, cytoplasm, cells, and bacteria), relating them (e.g., bacteria within cells), and then extracting morphological and intensity properties (e.g., area, intensity). The essential steps of the image analysis are described below. The full protocol used can be found in Supplementary Figure 5.
- Segment the nuclei using the DAPI signal, the cytoplasm using the DeepRed signal, and the bacteria using the mScarlet signal.
- Relate the nuclei and cytoplasm segmentation to create a mask for cells.
- Remove all border objects. Only cells that appear as full on the FOV will be considered (whole cells).
- Define output population Bacteria in whole cells by relating whole cells and bacteria.
- Define infected whole cells as having more than one bacteria region and non-infected whole cells as having less than one bacteria region.
- After all regions of interest are segmented and the populations are created, calculate and extract their properties. The following properties are extracted for each well.
- For Whole cells/ Infected whole cells/ Non-infected whole cells, extract
Number of cells (Sum per well)
Area [µm2] (Mean + Standard deviation per well)
Width-to-length cell ratio (Mean + Standard deviation per well) - For bacteria in whole cells, extract
Number of bacterial regions identified (Sum per well)
Intensity (Sum per well of each region's mean intensity)
Region's area (Sum per well)
- For Whole cells/ Infected whole cells/ Non-infected whole cells, extract
- After exporting the data and curating them in a spreadsheet, quantify intracellular load (Mycoload) according to the following formula:
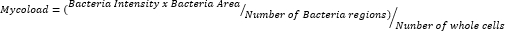
Z' factor assessment
Figure 2 depicts the data of one out of two experiments performed to calculate the Z' factor. The positive control was clarithromycin at 4 µg/mL. Both experiments yielded Z' factors of 0.64 and 0.62, meaning that the conditions and readout used for this assay can be applied to the screening assay that followed (Z' > 0.5). Nonetheless, the Z' factor was calculated for all the remaining experiments (infection assay) to control each experiment's performance.
As proof of concept of the designed HTS assay, a library of compounds intended for drug repurposing was tested. It comprises 1280 diverse and small molecules, 95% of which are both FDA- and EMA-approved drugs. These molecules offer high chemical and pharmacological diversity.
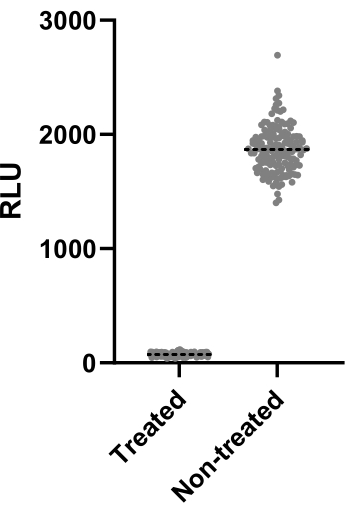
Figure 2: Z' factor assessment results. Mab at 2.5 x 105 CFUs/mL was incubated with and without clarithromycin at 4 µg/mL for 48 h at 37 °C. After the incubation period, the luminescence was measured to evaluate mycobacterial viability. The graph shows the individual luminescence values of treated and non-treated viable mycobacteria in one independent experiment. Please click here to view a larger version of this figure.
Library screening for hits
All compounds were tested at 6.66 µM against Mab. Due to the size of the library, the compounds were divided into four different microtiter 384-well plates (Figure 3A-D). The results were normalized for both solvent and the verified edge effect.
In this screening assay, thirty-three hits were identified, thirty of which significantly decreased luminescence emission, reducing mycobacterial viability (Figure 3). Interestingly, three compounds led to higher luminescence emission, possibly related to increased bacterial metabolism or proliferation (Figure 3). All 33 hits were carried on to the validation assay, including the three compounds that increased luminescence, to test if this profile would be kept.
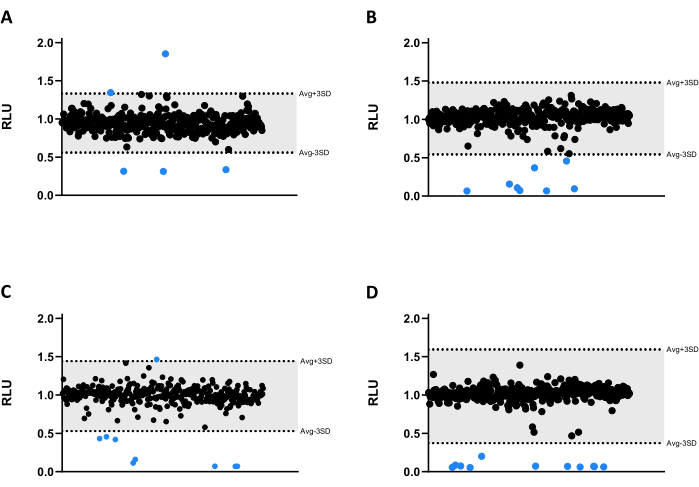
Figure 3: Library screening results. (A-D) Mab at 2.5 x 105 CFUs/mL was incubated with 1280 compounds at 6.66 µM for 48 h at 37 °C. After incubation, the luminescence was measured to evaluate mycobacterial viability. The graphs show the RLU of one independent experiment, presented as arbitrary units after solvent and edge effect normalization. The AVG RLU and its corresponding SD were calculated for each plate to determine a threshold (in grey; AVG ± 3SD). Round symbols represent a tested compound. In blue, any compound outside of that threshold is deemed a hit. Please click here to view a larger version of this figure.
Hit validation assay
The concentrations used for this assay were 13.3 µM, 6.66 µM, and 3.3 µM. Each compound was tested in duplicate to increase robustness.
Importantly, compounds 30, 31, and 32, previously associated with higher RLU (Figure 3A,C), kept this profile, with more than 200% of mycobacterial viability achieved in wells treated with compound 32 (Figure 4).
Compounds 20 and 33 were deemed inactive, as the mycobacterial viability is close to 100% in all concentrations tested (Figure 4).
Compounds 9 and 21 displayed similar inactivity at the two lowest concentrations; however, contrary to 20, they remain active at the highest, 13.3 µM, with compound 21 displaying higher potency (Figure 4).
Compound 3 also displays inactivity at the lowest concentration and an activity loss at 6.66 µM, albeit less than compound 21 (Figure 4). Only in the lowest concentration, compounds 2, 7, 8, 10, 16, 18, 24, and 28 displayed lower therapeutic potential. However, 2, 10, and 28 still lead to <50% of mycobacterial viability.
Globally, the remaining fourteen compounds were active in all concentrations tested.
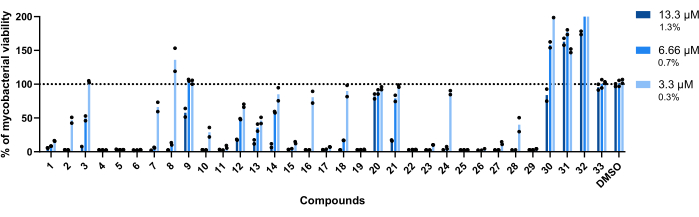
Figure 4: Hit validation assay results. Mab at 2.5 x 105 CFUs/mL was incubated with each previously identified hit at 13.3 µM, 6.66 µM, and 3.3 µM (1.3%, 0.7%, and 0.3% of DMSO, respectively) for 48 h at 37 °C. After the incubation period, the luminescence was measured to evaluate mycobacterial viability. The graph shows the percentages of treated viable mycobacteria relative to the non-treated mycobacteria of one independent experiment, with each compound tested in duplicate. Round symbols represent the duplicates for each compound. Bars represent the average of those duplicates. Please click here to view a larger version of this figure.
Infection assay
Out of thirty-three tested compounds, twenty-eight were selected for the infection assay (Figure 4). Compounds 7, 8, 9, 20, and 33 were not selected for this assay. While 9, 20, and 33 were left out due to their inactivity when validated (Figure 4), the first two were left out due to technical reasons. Nonetheless, these compounds were identified as rifampicin and linezolid, antibiotics already used to treat Mab infections12. All the compounds tested in the infection assay were identified and are listed in Table 1. The antimicrobial activity of the compounds against Mab-infected macrophages was assessed by using the intrinsic fluorescence of the bacteria as a readout.
| Compound | Name | Compound | Name |
| 1 | Sulfathiazole | 18 | Cefuroxime |
| 2 | Ciprofloxacin | 19 | Rifaximin |
| 3 | Cefotaxime | 21 | Cefdinir |
| 4 | Daunorubicin | 22 | Clarithromycin |
| 5 | Doxorubicin | 23 | Besifloxacin |
| 6 | Thiostrepton | 24 | Levofloxacin |
| 10 | Amikacin | 25 | Rifabutin |
| 11 | Moxalactam | 26 | Gatifloxacin |
| 12 | Sulfamethizole | 27 | Epirubicin |
| 13 | Sulfamonomethoxine | 28 | Pyrvinium pamoate |
| 14 | Cefoxitin | 29 | Moxifloxacin |
| 15 | Novobiocin | 30 | Troleandomycin |
| 16 | Cefmetazole | 31 | Lincomycin |
| 17 | Roxithromycin | 32 | Spiramycin |
Table 1: List of the compounds tested in the infection assay. The compounds validated in step 4 were tested in the infection assay (step 5).
The compounds' toxicity towards Mab-infected macrophages was the first parameter assessed. The threshold established to deem a compound as toxic or non-toxic was 85% of viable macrophages (Figure 5). Of the twenty-eight compounds tested, 4, 5, 6, 27, and 28 were deemed toxic. (Figure 5). Thus, these five compounds were excluded from the following intramacrophagic activity assessment.
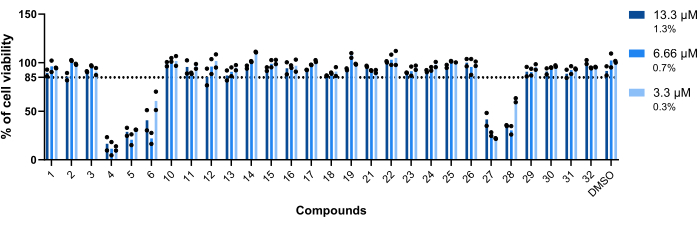
Figure 5: Hits' toxicity towards Mab-infected macrophages. BALB/c mice BMMs were infected with Mab (MOI=1) and incubated with each previously identified hit at 13.3 µM, 6.66 µM, and 3.3 µM (1.3%, 0.7%, and 0.3% of DMSO, respectively) for 48 h at 37 °C with 7% CO2. The cells were imaged in a high-content screening fluorescence microscope, using the number of nuclei (stained with DAPI) to measure cell viability. The graph shows the percentages of viable treated infected macrophages relative to the viable non-treated infected macrophages of two independent experiments. Round symbols represent the cell's viability for each assay. Bars represent the average of two independent experiments. Please click here to view a larger version of this figure.
To infer the intramacrophagic activity of the remaining twenty-three compounds against intracellular Mab (Figure 6), the Mycoload formula (previously explained in step 6) was used to obtain the percentage of mycobacterial viability normalized to non-treated infected macrophages. Most compounds lost their therapeutic potential against internalized mycobacteria (Figure 6) compared to the hit validation assay (Figure 4), as the number of hits reduced from twenty-five to six at the highest tested concentration. Strikingly, all three compounds that increased the viability of planktonic Mab compared to non-treated bacteria (30, 31, and 32; Figure 3 and Figure 4) displayed antimycobacterial activity against intracellular Mab, with compounds 30 and 32 presenting a significant statistical difference when compared to DMSO, even at 3.3 µM in the case of compound 32 (Figure 6). Mycobacteria treated with compounds 11 and 23 displayed viability <50% at 13.3 µM; however, this was not significantly different from the DMSO control (Figure 6). Compounds 21, 26, and 29 were potent enough at 13.3 µM to warrant a significant statistical difference, with 29 being the most active (Figure 6). Lastly, compounds 17, 22, and 25 were extremely potent in all tested concentrations against internalized mycobacteria. These were identified as roxithromycin, clarithromycin, and rifabutin, respectively (Table 1). Of the three compounds, clarithromycin was the most active against Mab, with mycobacterial viability never surpassing 10%, presenting a p-value <0.0001 in all concentrations tested compared to DMSO (Figure 6).
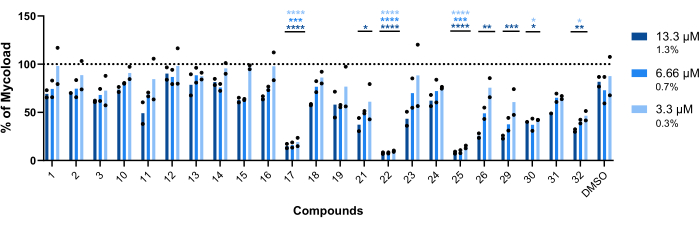
Figure 6: Hits' intramacrophagic activity against Mab-infected macrophages. BALB/c mice BMMs were infected with Mab (MOI=1) and incubated with each previously identified hit at 13.3, 6.66, and 3.3 µM (1.3, 0.7, and 0.3% of DMSO, respectively) for 48 h at 37 °C with 7% CO2. The cells were imaged in a high-content screening fluorescence microscope, and the fluorescent signal was used to calculate the Mycoload (see step 6). The graph shows the percentages of Mycoload found in treated infected macrophages relative to non-treated infected macrophages in two independent experiments. Statistics were performed using two-way ANOVA with Dunnet's multiple-comparison test; *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 compared to DMSO (solvent control). Asterisks follow the same color code for each concentration as the graph's legend. Round symbols represent the Mycoload for each assay. Bars represent the average of two independent experiments. Please click here to view a larger version of this figure.
Supplementary Figure 1: Example of a 384-well plate layout used in the Z' factor assessment. White – blank wells (liquid growth medium only); yellow – positive control (bacteria treated with an antibiotic); red – negative control (non-treated bacteria). Please click here to download this File.
Supplementary Figure 2: Example of a 384-well plate layout used in the library screening. White – blank wells (liquid growth medium only); green – solvent control; red – negative control (non-treated bacteria); blue – compounds to be screened. Please click here to download this File.
Supplementary Figure 3: Example of a 384-well plate layout used for hit validation. White – blank wells (liquid growth medium only); green – solvent control (in duplicates); red – negative control (non-treated bacteria); blue – compounds to be screened (in duplicates). Faded colors represent 1:2 serial dilutions. Please click here to download this File.
Supplementary Figure 4: Example of a 96-well plate layout used for an infection assay. White – blank wells (water to prevent evaporation); red – negative control (non-treated macrophages); yellow – positive control (macrophages treated with an antibiotic); green – solvent control; blue – compounds to be tested. Faded colors represent 1:2 serial dilutions. Please click here to download this File.
Supplementary Figure 5: Image analysis protocol. A detailed protocol of image analysis is used in this work, which can be adapted to open-source software. Please click here to download this File.
