Brightfield images showing the morphological changes of HMDM in response to stimuli for cell differentiation are shown in Figure 1. M1 polarized macrophages from experiments with HMDM exposed to IFNγ and LPS showed an elongated and spindle-like cell shape, as indicated by the black arrows in Figure 1 (middle panel). For comparison, the morphology of the M2 polarized macrophages after exposure of HMDM to IL-4 for 48 h were typically round and flat, as indicated by the black arrows in Figure 1 (far right panel).
The ability of differentiated HMDM phenotypes to release METs was visualized by live cell fluorescence imaging with SYTOX green, as presented in Figure 2. Figure 2A shows the control data obtained from each HMDM phenotype incubated for 24 h in the absence of any pro-inflammatory stimuli. In this case, there was very limited green staining, as was expected, given the cell impermeant nature of this stain. Figure 2B showed positive staining for METs, resulting from the exposure of M1 HMDMs to HOCl, PMA, IL-8, or TNFα. The METs are indicated by the white arrows, shown as green streaks, resulting from the strands of extracellular DNA. With HOCl, in addition to staining extracellular DNA, there was some green staining apparent in the cells. This cellular staining was also observed to some extent with the other stimuli and is believed to reflect loss of membrane integrity resulting from ET-independent cell death as a result of the treatment conditions.
Figure 2B also shows the corresponding experiments performed with M2 HMDMs, which were exposed to IL-4. In this case, there was no release of DNA from the cells, as indicated by the absence of the strands/streaks of extracellular DNA; though, there was some cellular uptake of green fluorescent dye with the HOCl and TNFα. For comparative purposes, Figure 3 shows representative data indicating MET release from THP-1 macrophages exposed to TNFα (50 ng/mL) for 4 h. In this case, it should be noted that a non-polarized population of cells was used, and the THP-1 monocytes were differentiated to macrophages by pre-treatment with PMA (50 ng/mL for 72 h) as described previously22.

Figure 1: Morphological changes of differentially polarized HMDMs. Representative brightfield images from non-differentiated and differentiated HMDMs (n ≥5). HMDMs were cultured with complete media containing human serum and glutamine for 8 days before priming to the M1 or M2 phenotype upon exposure to IFNγ and LPS or IL-4, respectively, for 48 h. Scale bar = 200 μm. Arrows indicate examples of cells showing morphological characteristics of M1 or M2 HMDMs. Please click here to view a larger version of this figure.
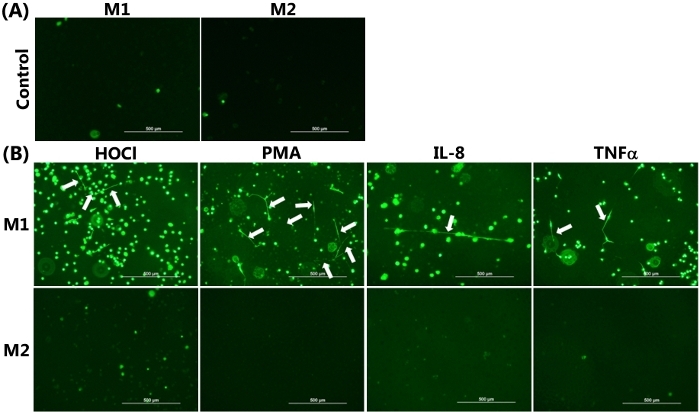
Figure 2: METs produced by M1 HMDMs following HOCl, PMA, IL-8, and TNFα stimulation. Representative images of SYTOX green stained HMDMs from (A) Non-stimulated M1 and M2 HMDMs incubated for 24 h in the absence of any inflammatory stimuli were used as the control, demonstrating the absence of MET release. (B) M1 and M2 HMDMs were treated with 1) HOCl (200 µM, 15 min), PMA (25 nM), or IL-8 (50 ng/mL) with incubation for 24 h or 2) TNFα (25 ng/mL) with incubation for 6 h to induce MET release. METs were visualized by the addition of SYTOX green, as indicated by white arrows in the upper panel from M1 HMDMs. No METS were seen in the corresponding experiments with M2 HMDMs. Data are representative of replicate culture wells from n ≥3 individual donors. Scale bar = 500 μm. Please click here to view a larger version of this figure.
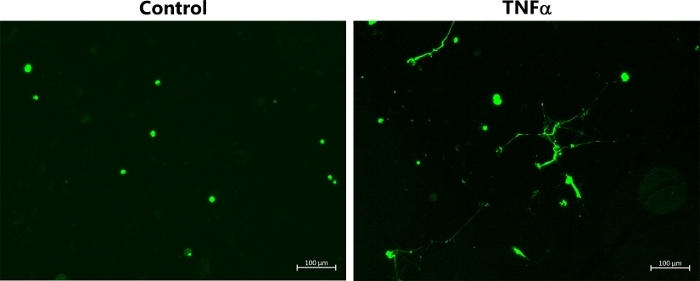
Figure 3: METs produced by non-polarized THP-1 macrophages following TNFα stimulation. Representative images of SYTOX green-stained TNP-1 macrophages, which were differentiated by pre-treatment with PMA (50 ng/mL for 72 h) before further incubation for 4 h in the absence or presence of TNFα (50 ng/mL) to induce MET release. METs were visualized by addition of SYTOX green. Data are representative of replicate culture wells from n ≥ 3 experiments. Scale bar = 100 μm. Please click here to view a larger version of this figure.
