This protocol describes a method for reproducible and robust production of endogenous yeast SNX-BAR complexes that can be used for downstream membrane remodeling assays (Figure 1). The construction of the yeast strain used for purification takes advantage of the efficiency of homologous recombination in budding yeast, allowing for modifications at the genomic loci of the targeted SNX-BARs (Figure 2). This design has two advantages, (i) as selection is not required to maintain the modifications, standard YP medium can be used, allowing for growth to a higher cell density and thus higher production of protein and (ii) the expression levels of the targeted SNX-BARs will be even, optimizing production of the heterodimer complex. Prior to addition of galactose, the targeted SNX-BARs will exhibit a null phenotype and thus may result in a growth defect or other known defects specific to the targeted SNX-BARs. Furthermore, growth in 2% raffinose and 0.1% glucose is slower than growth in 2% glucose. Therefore, the growth period prior to galactose induction may require optimization for each particular strain. To check for proper induction of SNX-BAR expression, a western blot against the TAP tag is likely required since protein levels of the SNX-BARs may not be detected via Coomassie stain (Figure 3). However, because only one member of the SNX-BAR complex has a tag, expression of the untagged protein(s) cannot be confirmed unless all steps of the purification are completed. After purification of the SNX-BAR heterodimer, the bands of the two SNX-BARs should appear to be in 1:1 stochiometric ratio and there should be little to no contaminating bands (Figure 4, lane 4). If there are additional contaminating bands, and the starting yeast strain is already protease-deficient, more protease inhibitor may be added during cell lysis. Furthermore, a second purification step using calmodulin resin may be performed.
When conducting membrane remodeling assays (Figure 5), the same preparation of liposomes and purified proteins must be used in the same experiment. If multiple purification preparations of protein are required to reach the desired concentration, combine all of the protein purified prior to conducting experiment. Liposomes should be made and used within the same day (Table 1). When conducting liposome sedimentation assays, it is critical that the purified protein is precleared using the same sedimentation conditions of 100,000 x g for 20 min immediately prior to incubating with liposomes as precipitated protein may skew results. Furthermore, the liposome pellet should stay intact after sedimentation.
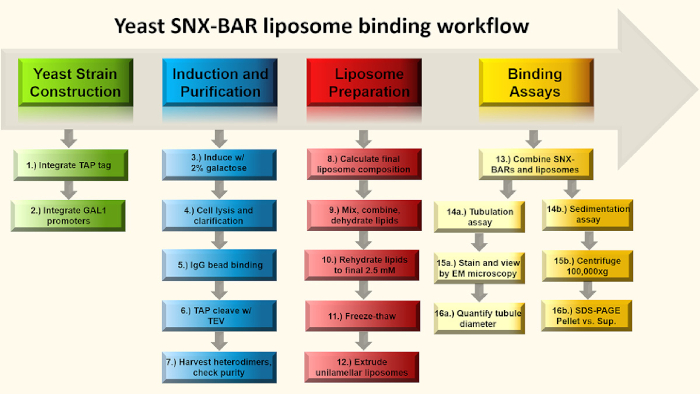
Figure 1: SNX-BAR binding assay flow chart. Briefly, in steps 1-2, we engineer GAL promoters into two SNX-BAR genomic loci, replacing each of the endogenous promoters and engineer a C-terminal TAP tag into one of the two SNX-BAR loci. Next, in steps 3-7, we induce the cells with galactose and purify the SNX-BAR heterodimers to homogeneity. In steps 8-12, we calculate and prepare unilamellar liposomes. Lastly, we can combine the SNX-BAR heterodimers with unilamellar liposomes and perform two assays; Step 14a-16a involve membrane tubulation and 14b-16a involve the sedimentation assay. See text for more details. Please click here to view a larger version of this figure.
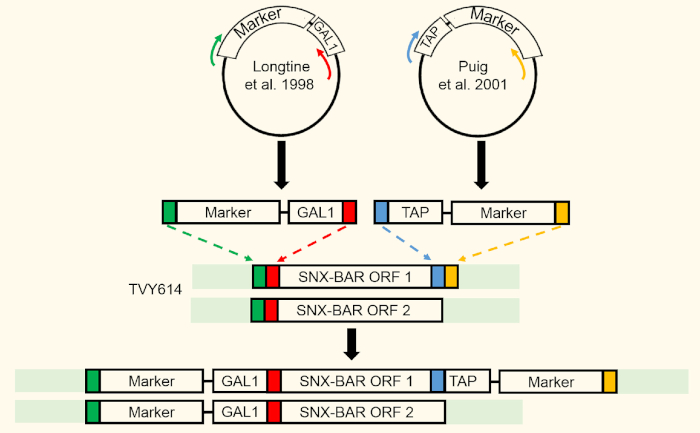
Figure 2: SNX-BAR integration strategy. Two SNX-BAR loci were targeted for GAL expression using homologous recombination. SNX-BAR ORF1 (Atg20) was additionally targeted to express a C-terminal TAP tag. Please click here to view a larger version of this figure.

Figure 3: Galactose induction verification. (A-B) In order to verify GAL induction of Atg20-TAP, we recommend an SDS-PAGE and western blot analysis of cell extracts induced with 2% galactose (A, B, lane 2) and uninduced (A, B, lane 1). (C) Western blot membrane is also stripped and probed with anti-pgk1 for a loading control. Please click here to view a larger version of this figure.
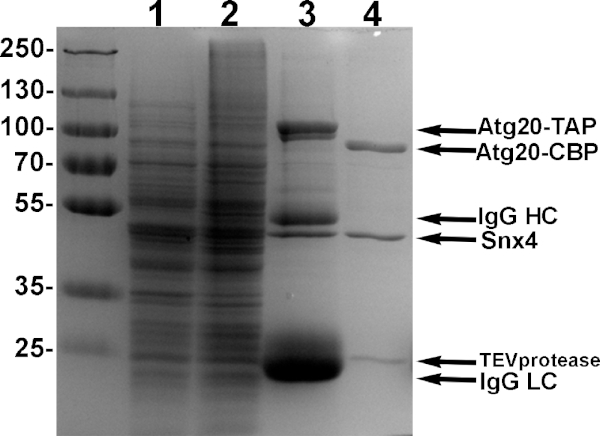
Figure 4: Example purification of Atg20-Snx4 heterodimers. Yeast cells engineered to express Atg20-TAP and Snx4 driven by galactose promoters were induced with 2% galactose, lysed and bound using IgG sepharose, and eluted with TEV protease. Samples from each step of purification is shown in 10% SDS-PAGE. Lane 1: induced supernatant of lysate. Lane 2: induced pellet of lysate. Lane 3: sample of bound proteins to IgG sepharose. Lane 4: TEV eluate of pure Atg20-Snx4 heterodimers from IgG sepharose. Please click here to view a larger version of this figure.
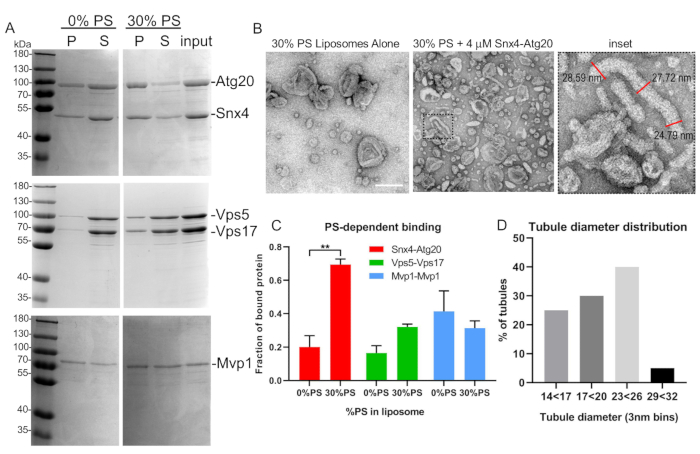
Figure 5: Representative liposome binding and tubulation assay. (A) SNX-BAR heterodimers, Atg20-Snx4 and Vps5-Vps17 from yeast, were expressed, purified, and bound to synthetic liposomes as described in text. Note that Mvp1 forms homodimers and was expressed in bacteria. (B) EM micrographs of Snx4-Atg20 liposome tubulation assay and tubule measurements (inset). (C) SNX-BAR binding to varying liposome compositions was quantified by densitometry. Graph indicates mean and standard error of the mean. **p < 0.002. (D) Tubule diameters were quantified and graphed as described in the text. Scale bar = 200 nm. Error bars represent two-way analysis of variance. Please click here to view a larger version of this figure.
| Typical Liposome Composition | DOPC | DOPS | Ergosterol | PI3P-C16 |
| MW | 786.1 | 810.0 | 396.7 | 957.0 |
| mol fraction | 49% | 30% | 20% | 1% |
| Stock mM | 32.0 | 12.0 | 25.0 | 1.0 |
| Mass (mg) | 385.2 | 243.0 | 79.3 | 9.6 |
| Concentration (mg/mL) | 25.2 | 9.7 | 9.9 | 1.0 |
| Volume to RXN(mL) | 15.3 | 25.0 | 8.0 | 10.0 |
Table 1: Liposome recipe. Synthetic liposomes were prepared using a combination of DOPC, DOPS, ergosterol, and PI3P. We calculate the final concentration to 1 mol of lipid. Our standard composition includes 20% ergosterol, 1% PI3P, DOPS (up to 30%), and varying amounts of DOPC. Table includes a typical formulation for 400 µL of 30% DOPS.
