All novel compounds in this study were characterized by 1H and 13C NMR spectroscopy and high resolution mass spectrometry. Previously reported compounds were characterized by 1H NMR spectroscopy. NMR data for representative compounds are described in this section.
The 1H NMR spectrum of tricarbonyl(tropone)iron is shown in Figure 3. The protons of the η4-diene ligand give rise to the signals at 6.39 ppm (2 H), 3.19 ppm, and 2.75 ppm. The protons from the uncomplexed double bond appear at 6.58 and 5.05 ppm.
The progress of the aza-Michael addition is monitored via 1H NMR by observing the disappearance of the signals from the uncomplexed double bond and a characteristic change in the chemical shift of the two furthest downfield η4-diene protons from around 6.4 ppm to two well separated signals that typically appear between 5.3 and 6.0 ppm (see Figure 3 and Figure 4). Furthermore, the aza-Michael adduct features signals corresponding to the two diastereotopic methylene protons (adjacent to the ketone within the seven-membered ring), which typically appear between 1.5 and 2.5 ppm.
Direct aza-Michael additions to tricarbonyl(tropone)iron generally proceeded in 60-95% yield, depending on the amine substrate (see Discussion). Secondary cyclic amines tend to give somewhat higher yields than primary aliphatic amines, possibly due to a greater resistance to decomposition during purification.
1H NMR data for the cationic complex (in CD3CN) is shown in Figure 5 and features seven distinct multiplets. It should be noted that the complex decomposes over time in CD3CN. However, the dried solid tetrafluoroborate complex can be stored indefinitely under ambient conditions. Figure 6 shows 1H and 13C NMR data for the o-toluidine adduct 3, prepared via the cationic complex 2 (Figure 1), which contains the same features described above for the phenethylamine adduct 4.
Figure 7 shows 1H and 13C NMR spectra of tert-butyl carbamate 5. The 1H NMR spectrum is characterized by its broad peaks, caused by slow rotation of the carbamate C-N bond relative to the NMR time scale. In addition, the presence of the tert-butyl carbamate is evident from the large singlet at 1.5 ppm from the tert-butyl protons, as well as the signal at 154.3 ppm in the 13C NMR spectrum corresponding to the carbonyl carbon of the carbamate group.
Upon decomplexation of the diene from the iron, the most notable aspect of the 1H NMR spectrum (Figure 8) is the presence of four signals between 5.75 and 6.75 ppm, corresponding to the protons from the uncomplexed diene.
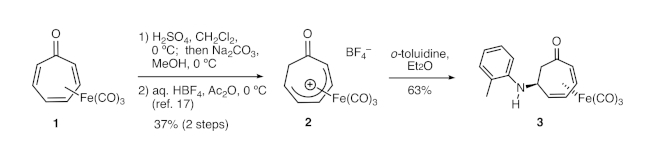
Figure 1. Synthesis of 3 from tricarbonyl(tropone)iron via cationic complex 2. Tricarbonyl(tropone)iron is converted to cationic complex 2 in two steps, which was followed by nucleophilic addition of ortho-toluidine to the complex. Please click here to view a larger version of this figure.
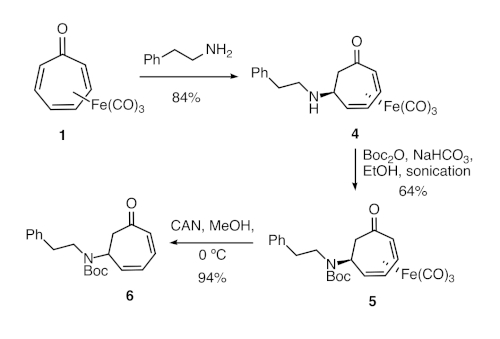
Figure 2. Synthesis of formal tropone aza-Michael adduct 6. Direct aza-Michael reaction of tricarbonyl(tropone)iron and phenethylamine was followed by amine protection and oxidative demetallation. Please click here to view a larger version of this figure.
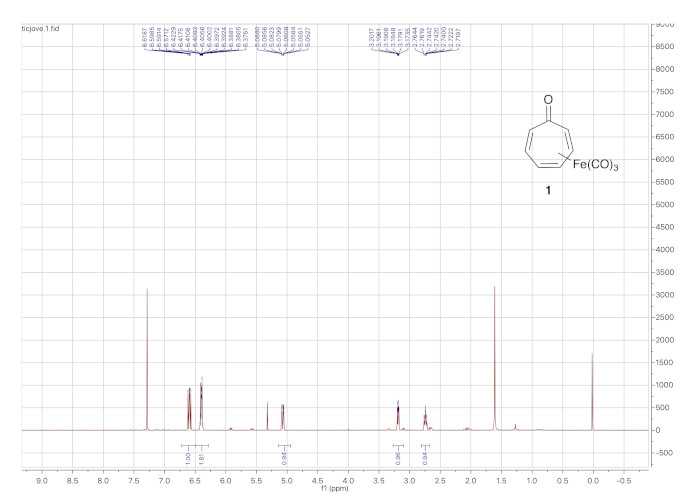
Figure 3. 1H NMR spectrum (solvent: CDCl3) of tricarbonyl(tropone)iron 1. The peaks at 6.59 ppm and 5.05 ppm correspond to the uncomplexed alkene hydrogens while those 6.39 ppm (2H), 3.19 ppm, and 2.75 ppm arise from the iron-complexed diene. Please click here to view a larger version of this figure.
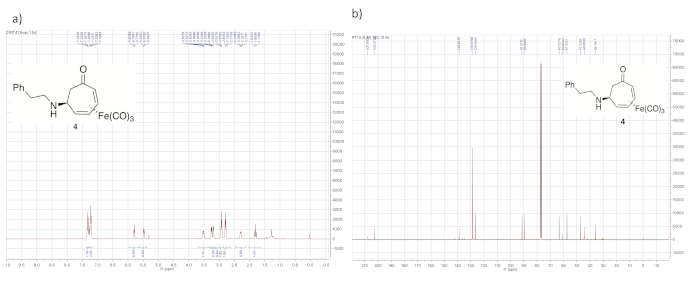
Figure 4. Spectral data for iron complex 4. (a) 1H NMR spectrum; (b) 13C NMR spectrum (solvent: CDCl3). Notable peaks in the 1H NMR spectrum include those from the iron-complexed diene (5.75, 5.48, 3.30, and 3.20 ppm) and the diastereotopic α-methylene protons (2.30 and 1.70 ppm). Please click here to view a larger version of this figure.
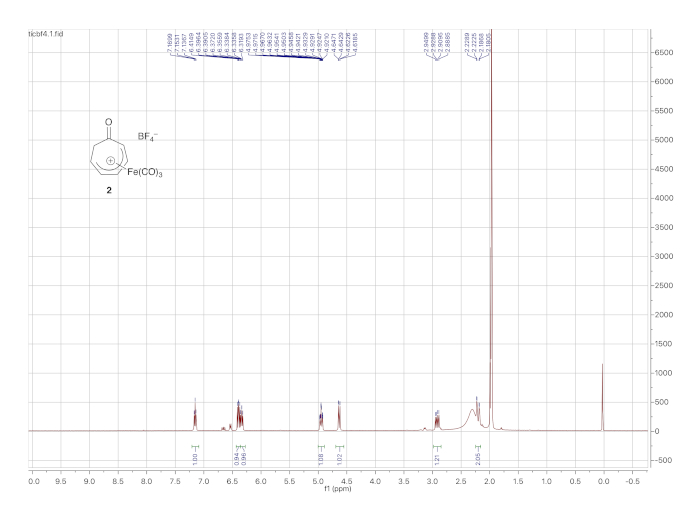
Figure 5. 1H NMR spectrum (solvent: CD3CN) of cationic iron complex 2. The most notable difference from the 1H NMR spectrum of 1 (the precursor to 2) is the signals arising from the diastereotopic α-methylene protons (2.85 and 2.23 ppm). Please click here to view a larger version of this figure.
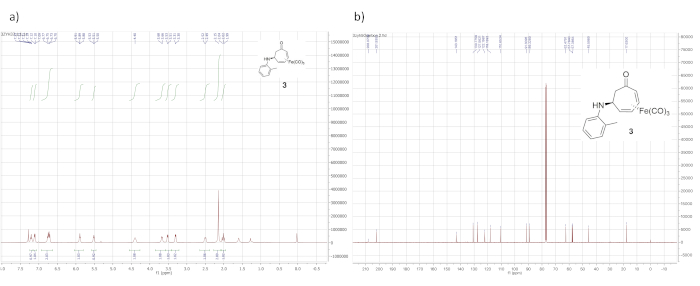
Figure 6. Spectral data for iron complex 3. (a) 1H NMR spectrum; (b) 13C NMR spectrum (solvent: CDCl3). Similar to the 1H NMR spectrum of 4, the 1H NMR spectrum of 3 is characterized by signals arising from the iron-complexed diene (5.89, 5.51, 3.53, and 3.30 ppm) and the diastereotopic α-methylene protons (2.50 and 2.02 ppm). Please click here to view a larger version of this figure.
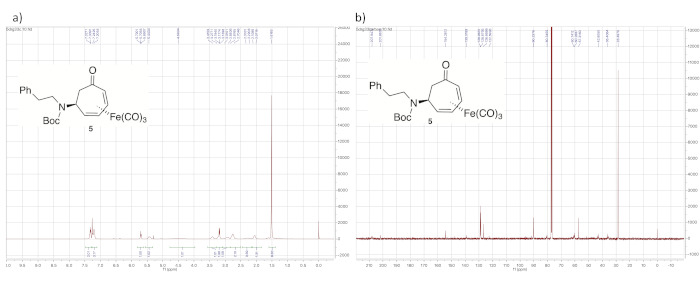
Figure 7. Spectral data for tert-butyl carbamate 5. (a) 1H NMR spectrum; (b) 13C NMR spectrum (solvent: CDCl3). The signal corresponding to the protons of the tert-butyl group of the carbamate appear at 1.52 ppm. Many signals also show characteristic broadening. Please click here to view a larger version of this figure.
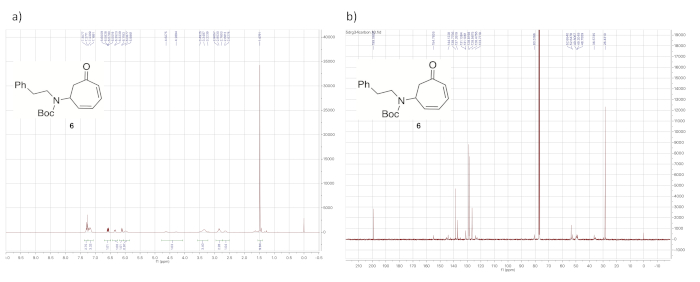
Figure 8. Spectral data for demetallated diene 6. (a) 1H NMR spectrum; (b) 13C NMR spectrum (solvent: CDCl3). The most notable aspect of the 1H NMR spectrum compared to those of the iron complexes in Figure 4a, Figure 6a, and Figure 7a is that all of the signals corresponding to the diene protons now appear above 5.75 ppm (6.57, 6.34, 6.10, and 5.99 ppm). Please click here to view a larger version of this figure.
