1. Cloning of expression vectors and transformation of an auxotrophic production strain
Herein, the genes for nisin biosynthesis, namely nisABC, have been taken from L. lactis and transferred into T7-based plasmid expression vectors. Full DNA sequences of nisABC can be found in GenBank entry X6830734. The gene for the precursor peptide (nisA) has been placed on a pET-3a vector that confers ampicillin resistance. Genes for the dehydratase (nisB) and cyclase (nisC) have been placed on vector pRSFDuet-1, as reported earlier35, which confers kanamycin resistance.
NOTE: For nisA, the codons of the last four amino acids (ASPR) of the leader sequence were mutated to encode VSLR36 to render the proline residue in the core peptide unique and to ensure proper prepeptide processing by NisP. At the N-terminus, a hexa-histidine tag flanked by linker residues was added for purification purposes (see Figure 1).
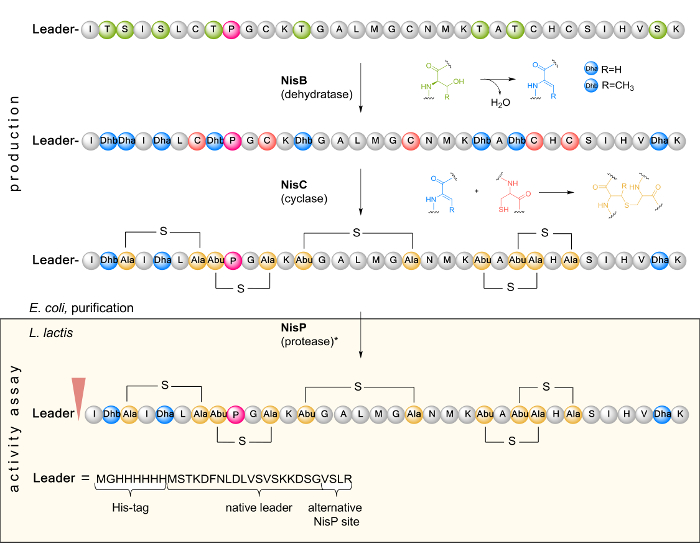
Figure 1. Schematic representation of the biosynthesis and PTM of NisA in E. coli as well as leader cleavage by the L. lactis indicator strain in a subsequent activity assay. In the first step, the inactive linear prenisin (composed of a leader and a core peptide region containing a unique proline (pink) at position 9) encoded by nisA is ribosomally synthesized. Next, prenisin is posttranslationally modified by dehydration of serine and threonine residues to dehydroalanine (Dha) and dehydrobutyrine (Dhb) as catalyzed by NisB. The cyclase NisC forms the thioether bridges via Michael addition of cysteine sulfhydryl groups with Dha or Dhb. The inactive modified prenisin is purified from E. coli and tested for antimicrobial activity. Here, it is transported into the cell of the Gram-positive L. lactis indicator strain. The leader is cleaved by protease NisP (as indicated by an arrowhead) to release fully active nisin. It can also be removed in vitro by treatment with trypsin (*). Please click here to view a larger version of this figure.
- Use standard heat-shock protocol37 or electroporation38 to transform the proline auxotrophic E. coli strain (see above) with the plasmids pET-3a nisA(VSLR) and pRSFDuet-1 nisBC.
- Pipette 25-100 µL of transformed cell suspension onto agar plates containing ampicillin, kanamycin and 1% (w/v) glucose. Use a plate spreader or glass beads to spread the solution evenly on the plates.
- Incubate plates overnight at 37 °C.
- The following afternoon, use a single colony to inoculate 10 mL LB medium containing ampicillin, kanamycin and 1% (w/v) glucose in a 50 mL flask.
- Shake culture overnight (12-16 h) at 37 °C and 200 rpm.
- Take 250 µL sterile 80% glycerol and 550 µL culture, mix well in a 2 mL tube and store as frozen cell stock at 80 °C.
2. New minimal medium (NMM) preparation
NOTE: This protocol uses NMM20 as a chemically defined liquid bacterial growth medium. Also, it is advised to follow the order of preparation strictly. Otherwise, precipitation can occur. For amino acid forms differing from those listed in the Materials table (e.g., hydrochlorides), check solubility. NMM19 contains 19 amino acids except for the cAA to be replaced (here, proline) by the ncAA analog. See Table 1 for the final ingredient concentrations. Depending on the bacterial strain used for production, biotin and thiamine may be optional.
- Preparation of the amino acid mixture
- Dissolve 0.5 g Phe, Trp and Tyr in 100 mL ddH2O with the addition of a few drops of concentrated HCl until dissolution.
- Weigh out 0.5 g of each of the remaining 16 amino acids. Mix with 22 mL of 1 M KH2PO4 and 48 mL of 1 M K2HPO4. Add ddH2O to ~800 mL. Stir until the solution becomes clear.
- Add the dissolved Phe, Trp and Tyr and adjust the volume of the solution to 1 L with ddH2O.
- Sterilize the amino acid mixture by vacuum filtration with a bottle top filter unit.
- Stock solutions for NMM19
- First, prepare 1 M stock solutions of the following components: (NH4)2SO4, KH2PO4, K2HPO4, MgSO4 and a 5 M stock solution of NaCl. Sterilize by autoclaving.
- Prepare 50 mL stocks of D-glucose (1 M), CaCl2 (1 g/L), FeCl2 (1 g/L), thiamine (10 g/L), biotin (10 g/L) and trace elements (CuSO4, ZnCl2, MnCl2, (NH4)2MoO4; 1 mg/L). Sterilize each by filtration with a syringe filter.
- NMM19 preparation
- Mix all stock solutions to obtain a final concentration of 7.5 mM (NH4)2SO4, 1.7 mM NaCl, 22 mM KH2PO4, 50 mM K2HPO4, 1 mM MgSO4 and 20 mM D-glucose, 50 mg/L amino acid mix, 1 µg/L CaCl2, 1 µg/L FeCl2, 10 µg/L thiamine, 10 mg/L biotin and 0.01 µg/mL trace elements.
3. Expression of Recombinant Nisin with Incorporation of Proline Analogs by SPI
In this section, recombinant expression of the prepeptide (here: nisA) and PTM genes (here: nisBC) is performed. First, cells are grown in the presence of all cAAs, since LB complex medium is used. Glucose is added to repress the target gene expression at background level, which could otherwise lead to the production of wild-type peptide (here: nisin) due to leakiness of the promoters. Only after the target cAA (here: proline) is depleted, the ncAA is added and target gene expression is induced in chemically defined medium. Incubation of liquid cultures should be performed in suitable flasks with aeration (e.g., 500 mL in a 2 L Erlenmeyer flask at 200 rpm).
- Using a sterile pipette tip, start a fresh overnight culture from frozen cell stock or fresh colony (see step 1). Use 25 mL LB medium containing ampicillin, kanamycin and 1% (w/v) glucose and incubate overnight (12-16 h) at 37 °C and 200 rpm.
- Inoculate 1 L of sterile fresh medium with 10 mL overnight culture (1% v/v) and incubate at 37 °C and 200 rpm until OD600 = 0.5.
- Centrifuge at 4 °C for 15 min at 4,500 x g.
- Pour off the supernatant and resuspend the pellet with 20 mL NMM19 (prepared in step 2.3) containing antibiotics and 1% (w/v) glucose. Centrifuge at 4 °C for 10 min at 4,500 x g.
- Resuspend cell pellet in 500 mL of the same medium and incubate at 30 °C and 200 rpm for 1 h.
NOTE: At this step, the cAA depletion (here, proline) takes place. - Divide the culture into equal parts (one for each ncAA). Induce each culture with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) and supply 1 mM proline analogs (4S/R-fluoroproline, 4S/R-hydroxyproline or 4S/R-methanoproline).
NOTE: As control, one culture can be supplied with 1 mM proline, resulting in wild-type peptide production. - Incubate overnight (12-16 h) at 28 °C and 200 rpm.
- Centrifuge the cell cultures in 50 mL tubes at 4 °C for 20 min at 5,000 x g. Pour off the supernatant and store pellets at -80 °C until purification.
4. Isolation and Purification of His-tagged Nisin Analogs
Peptides are purified under denaturing conditions with guanidine hydrochloride (GuHCl)39, a strong denaturant.
Caution: GuHCl is harmful if swallowed or inhaled and causes skin and serious eye irritation. Wear eye protection and gloves.
- Prepare 250 mL of binding buffer (5 M GuHCl, 300 mM NaCl, 25 mM Tris, pH 7.4), wash buffer (300 mM NaCl, 25 mM Tris, 25 mM imidazole, pH 7.4) and elution buffer (300 mM NaCl, 25 mM Tris, 250 mM imidazole, pH 7.4). For these, transfer the solids into a 250 mL bottle and fill up to 200 mL with ddH2O. Mix well and adjust the pH to 7.4 with 1 M NaOH or HCl. Then, fill up to 250 mL with ddH2O. Filter all buffer solutions using a bottle top filter unit.
- Cell lysis
Here, a sonicator (with 200 W maximum high frequency (HF) output) is used; note that power settings needed for cell disruption may differ for other instruments. All steps are performed on ice. Alternatively, chemical cell lysis, a liquid homogenizer or a French press can be used.- Add 12 mL binding buffer to each centrifuge tube (from step 3.8) and resuspend by vortexing.
- Submerge the tip of the sonicator probe into the cell suspension. Set sonicator at 40% amplitude with pulse of 1 s on / 5 s off for 15 min.
NOTE: Clean the sonicator tip between samples to avoid carryover. Wipe the sonicator probe with 70% ethanol. - Centrifuge the lysed cell suspension at 4 °C for 40 min at 15,000 x g to pellet cell debris. Transfer the supernatants to a new reaction tube.
- Affinity chromatography
For immobilized metal ion affinity chromatography (IMAC)40, a peristaltic pump or an FPLC system can be used with a 1 mL cartridge (here filled with Ni-NTA resin). For preparation of buffers, see step 4.1.
NOTE: IMAC purification is feasible since the produced recombinant peptide carries an N-terminally His-tagged leader, which is removed in step 6 by leader peptidase NisP, releasing mature nisin. Perform purification at room temperature or at 4 °C. Use a flow rate of 1 mL per minute if applicable for the IMAC cartridge.- First, wash the cartridge with 5 column volumes (cv) of ddH2O to remove storage buffer.
- Equilibrate with 10 cv of binding buffer.
- Process cell lysate (step 4.2) using a syringe filter to remove particles, then apply to the cartridge.
- Wash with 15 cv of wash buffer in order to remove nonspecific and unbound material.
- Elute with 10 cv of elution buffer and collect 1 mL fractions in 1.5 mL tubes. Store the fractions at 4 °C for short term (up to 3 days) or at -20 °C for longer terms.
- For storage, wash the cartridge with 10 cv of ddH2O followed by 5 cv of 20% ethanol.
5. LC-ESI-TOF Mass Spectrometric Analysis of Nisin Analogs
NOTE: See Materials table for example instrumentation for liquid chromatography coupled with electrospray ionization time-of-flight mass spectrometry (LC-ESI-TOF-MS).
- Perform HPLC separation of 15-20 µL peptide solution (prepared in step 4.3) on a C5 column with a mobile phase of water (A) and acetonitrile (B) both supplemented with 0.1% formic acid and a gradient from 5-80% B over 20 min. For mass spectrometry (MS), use elution after 5 min.
NOTE: Depending on peptide content and affinity to the HPLC column, sample volumes and separation may need optimization. - Use appropriate software to deconvolute the measured mass spectra and calculate the different peptide charge states41. Compare the observed peptide species mass to the calculated wild-type mass altered by the cAA → ncAA substitution. Take into account that the linear prepeptide is posttranslationally modified by eight dehydrations (-8 H2O) and five cyclizations (see Figure 1).
NOTE: Using sodium-containing buffers, MS analysis in positive mode can show sodium adducts. These become visible as additional peaks with higher deconvoluted mass (for each sodium adduct, the observed deconvoluted mass is 22.99 Da higher). To remove these adducts, HPLC purification42 or extensive dialysis43 can be performed.
6. Antimicrobial Activity Test
- Preparation of GM17-agar plates under sterile conditions
- Prepare an overnight culture of the indicator strain L. lactis NZ9000 carrying plasmid pNG nisPT44 at 30 °C in M17 broth45 with 1% (w/v) glucose (=GM17) and 5 µg/mL chloramphenicol.
- Measure OD600, inoculate fresh medium to OD600 = 0.1 and incubate until OD600 = 0.4-0.6. Then place the flask on ice.
Note: Each OD600 measurement will consume culture volume. Keep in mind that for each assay agar plate, 1 mL bacterial culture will be needed. If required, scale up the liquid culture volume accordingly. - For 1.5% agar, weigh out 4.5 g agar in a glass media bottle. Add 300 mL ddH2O, mix, and autoclave.
- Prepare 2x M17 broth (two-times concentrated) in 300 mL ddH2O and autoclave.
- Mix 25 mL 2x M17 broth containing 10 µg/mL chloramphenicol and 2% glucose with 1 mL L. lactis preculture (4% v/v).
- Add 25 mL molten 1.5% agar (freshly autoclaved or heated in a microwave).
NOTE: Prior to this, let the bottle cool to the touch (around 50 °C). This is necessary since L. lactis is a mesophilic organism sensitive to high temperatures. - Pour the solution into a large petri dish. Dry plates for 10-15 min.
- Sterilize the ends of a glass Pasteur pipette by flame. Wait for it to cool down, then use the wide end to create holes in the solidified GM17-agar.
- Sample preparation
- Take 1 mL of the E. coli expression cultures (created in step 3.7) in a labelled 1.5 mL tube and centrifuge for 3 min at 7,000 x g. Aspirate the remaining medium and resuspend the cell pellet in 500 µL Na-P (50 mM sodium phosphate buffer, pH 7.4 made from 0.5 M sodium dihydrogen phosphate and 0.5 M disodium hydrogen phosphate).
- Sonicate the samples on ice (compare step 4.2.2). Submerge the tip of the sonicator probe into the cell suspension. Set sonicator at 30 % amplitude with pulse of 1 s on and 5 s off for 3 min.
- Centrifuge the cell lysate for 10 min at 13,000 x g to pellet cell debris. Transfer the supernatant to a new reaction tube on ice.
- Dilute and normalize the cell extract supernatants to 1 mL OD600 = 0.6, relative to the harvested cell density, with Na-P.
- Activity test
- Add 40 µL of each normalized sample into a hole of the indicator agar plate (Figure 3). Use chloramphenicol at 400 µg/mL as antibacterial control compound. Use elution buffer as a negative control. Wait until all samples are diffused into the agar. Incubate the plate overnight at 30 °C.
- Take pictures of the agar plates using a flatbed scanner or digital camera. Growth inhibition halo sizes can be measured by hand or using ImageJ46.
7. Fluorescence Microscopy
In order to observe the effect of AMPs on bacterial cells, light and fluorescence microscopy can be used. Note that the nisin mode of action relies on the destabilization and the formation of pores in bacterial membranes6. Here, Nile red is used to stain the bacterial cell membrane, which becomes scattered and aggregated upon cell lysis.
NOTE: See Materials table for example instrumentation. Amounts of added AMP solution can be adjusted depending on peptide concentration and bioactivity.
- Cell preparation
- Prepare 10 mM Nile red stock solution in dimethyl sulfoxide (DMSO).
- Grow L. lactis indicator strain to OD600 = 1.0 as in step 6.1.1-6.1.2.
- Centrifuge 1 mL culture for 3 min at 4 °C and 5,000 x g.
- Discard supernatant, resuspend in 1 mL phosphate-buffered saline (PBS)47.
- Centrifuge and resuspend again.
- Add 1 µL Nile red stock solution, mix gently.
- Microscopic Image Acquisition
- Add 30 µL of cell preparation on a cover slide while exciting at 520 nm.
- Set acquisition time 0.2 s, kinetic series 0.1 Hz, series length 200 images.
- Add 0.3 – 1.5 µL of cell lysate or IMAC sample (from step 6.2.4 or 4.3.5, respectively). For IMAC samples, elution buffer can be used as a negative control.
- Monitor and record fluorescence emission at λ ≥ 560 nm.
- Data analysis
- Microscopy image sequences are stored as movie files (.avi). Single images are analyzed with ImageJ46.
This protocol is designed to enable the production of ncAA-modified nisin variants with residue-specific incorporation of proline analogs by the SPI method. Previously, the feasibility and yields of 24 mg/L were reported for recombinant production of fully modified wild-type nisin39. Using the SPI method, target peptide/protein yields are frequently good and can reach quantities close to wild-type production48. As first experiments, recombinant wild-type RiPP production should be tested in the chosen auxotrophic host. Here, the proline-auxotrophic E. coli MG1655 ΔproBA::frt ΔproC::frt (DE3) was used as host strain. For the incorporation of ncAAs, cultivation and induction timing as well as medium composition and temperature can be optimized towards maximum peptide yield.
Antimicrobial activity assay
Wild-type recombinant nisin production and purification were performed following the protocol above. In this case, proline was used instead of its ncAA derivatives at step 3.6. An antimicrobial activity assay was used to verify RiPP production and to compare the antimicrobial activity before and after purification. For the activity assay, elution fractions and IMAC flow-through were used directly and tested against the Gram-positive L. lactis indicator strain (Figure 2). As this strain expresses NisP, the nisin variants contained in E. coli cell lysates or purified peptide samples, respectively, become activated by proteolytic cleavage of the leader peptide. Evidently, the flow-through showed growth-inhibitory activity. This can be explained by bioactive material not binding to the IMAC column. The tested elution fractions all showed increased activity compared to the unpurified sample, indicating a concentration of the His-tagged peptide by IMAC. Note that the elution buffer (as negative control) did not influence the growth of L. lactis in this assay.
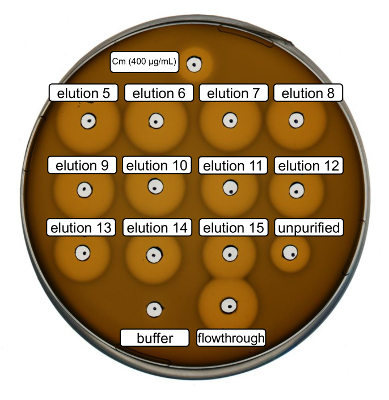
Figure 2. Antimicrobial activity test after IMAC purification of recombinantly produced wild-type nisin. Elution fractions 5 to 15 and the flow through of IMAC purification were tested in comparison with the unpurified cell lysate (diluted for OD600 normalization) against the L. lactis indicator strain. The size of growth inhibition halos indicates antimicrobial activity. Chloramphenicol at a concentration of 400 µg/mL was used as a positive control and the IMAC elution buffer as negative control. Please click here to view a larger version of this figure.
To prove the antimicrobial activity of recombinantly produced nisin variants containing six different proline analogs, an activity assay was performed to test the inhibition of the L. lactis indicator strain. Figure 3 shows growth inhibition for five of the six samples produced using proline analogs. The best results (as judged from halo sizes) were observed for the incorporation experiments of analog (4R)-fluoroproline, (4R)-hydroxyproline and (4S)-methanoproline. Comparing the growth inhibition halo size to the wild-type nisin produced and tested in parallel, all three nisin variants showed similar inhibition strength. However, the halo size alone cannot be used to asses a specific activity, since the concentration of AMP was not determined. Therefore, the assays serve only to test qualitatively whether the antimicrobial activity of the resulting nisin variants is preserved or lost. To determine the specific activity, the concentration of the nisin variants must be quantified (see Discussion).
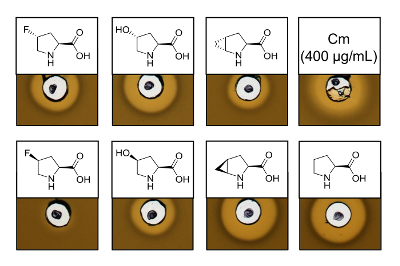
Figure 3. Antimicrobial activity assay of cell lysates containing nisin variants produced via SPI with proline analogs. Comparison of nisin variants with recombinant wild-type samples. All samples were OD600-normalized after cell lysis relative to the harvested cell culture density. Halos indicate bioactivity in form of indicator strain growth inhibition. First row from left to right: (4R)-fluoroproline, (4R)-hydroxyproline, (4R)-methanoproline and chloramphenicol (400 µg/mL; antimicrobial positive control). Second row: (4S)-fluoroproline, (4S)-hydroxyproline, (4S)-methanoproline and proline (wild-type control). Note the chemical nomenclature; e.g., (4R)-fluoroproline is also referred to as trans-4-fluoroproline. Please click here to view a larger version of this figure.
LC-ESI-TOF mass spectrometry
Subsequent to IMAC purification, the incorporation of the ncAAs into nisin was analyzed by LC-ESI-TOF mass spectrometry. Figure 4 shows the deconvoluted mass spectra of a nisin variant containing (4R)-fluoroproline. This variant was IMAC purified as described above and afterwards analyzed by LC-ESI-TOF mass spectrometry, so it still carried the leader. The main peak in Figure 4A corresponds to the modified nisin containing (4R)-fluoroproline with a deconvoluted mass of 6883.18 Da (calculated mass 6882.05 Da, calculated mass of the corresponding wild-type peptide with proline at position 9 is 6864.06 Da). The two peaks with lower abundance and higher mass correspond to sodium adducts as indicated. Figure 4B shows differently charged species of the main compound as found by the deconvolution algorithm. For example, the peak at 1148.11 m/z corresponds to the six-fold charged species ([M+6H]6+).
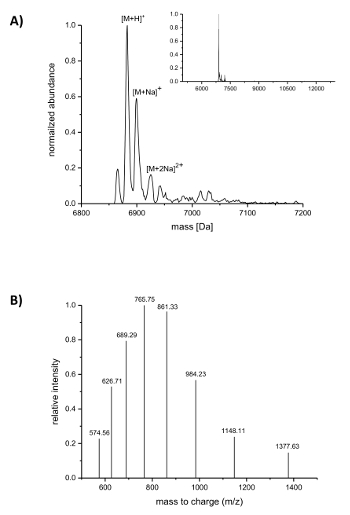
Figure 4. LC-ESI-TOF mass spectrometry of IMAC-purified recombinant nisin variants containing (4R)-fluoroproline. (A) Deconvoluted mass spectrometry chromatogram (zoomed out in the inset) for the nisin variant (still carrying the leader) with (4R)-fluoroproline (expected masses (Da): [M+H]+ = 6882.05, [M+Na]+ = 6904.03, [M+2Na]2+ = 6926.02). (B) Compound spectra for species [M+H]+. Expected masses (Da): [M+5H]5+ = 1377.41, [M+6H]6+ = 1148.01, [M+7H]7+ = 984.15, [M+8H]8+ = 861.26, [M+9H]9+ = 765.67, [M+10H]10+ = 689.21, [M+11H]11+ = 626.64, [M+12H]12+ = 574.50. Please click here to view a larger version of this figure.
Fluorescence microscopy
Antimicrobial activity of recombinant nisin and its ncAA-containing variants can also be shown by direct observation of the L. lactis indicator strain via fluorescence microscopy. Nile red, a highly hydrophobic fluorescent dye, was used to stain the bacterial cell membrane. Figure 5 shows the qualitative change of the aggregation state of the cell culture and single cell morphology. Cells were stained with Nile red and deposited on a microscopy cover slide. The upper row shows the cells directly at the beginning when buffer, recombinant wild-type nisin, or nisin containing (4R)-fluoroproline, or (4R)-hydroxyproline were added. The lower panel shows the corresponding images after 20 minutes of incubation.

Figure 5. Fluorescence microscopy of recombinant nisin effects on Gram-positive cells. Microscopic images of 30 µL L. lactis indicator strain (OD600 = 1) marked with Nile red were taken before (upper panel) and after (lower panel) 20 min incubation with 1 µL buffer (A), 0.3 µL recombinant wild-type nisin (B) and 0.6 µL nisin variants containing (4R)-fluoroproline (C) and (4R)-hydroxyproline (D). Blue circles mark regions with aggregated or deformed cells, blue arrows point to regions where diffusion of fluorescent membrane fragments can be observed. Please click here to view a larger version of this figure.
Figure 5A shows that the general appearance of the cells did not change within 20 minutes of observation. Only the number of cells that deposited during the time rose and, therefore, a larger amount of cells is visible within the 80 µm x 80 µm of the observed region. Figure 5B shows that the Gram-positive cells appeared aggregated and blurry (marked with blue circles) after 20 minutes of exposure to wild-type nisin, even when low amounts (here, 0.3 µL IMAC elution) were added. In addition, light material diffused from the cells into the buffer, indicating that membrane fragments marked with Nile red were mobilized during the timeframe (marked by blue arrows). These findings indicate cell lysis as it was shown to occur upon treatment with nisin6,49. Similar effects were observed after incubation with the nisin variant containing (4R)-fluoroproline (Figure 5C) and nisin containing (4R)-hydroxyproline (Figure 5D) both showing large amounts of distorted and aggregated cells after 20 minutes, in marked contrast to the control sample (Figure 5A).
| Component | Concentration |
| (NH4)2SO4 | 7.5 mM |
| NaCl | 8.5 mM |
| KH2PO4 | 22 mM |
| K2HPO4 | 50 mM |
| MgSO4 | 1 mM |
| D-Glucose | 20 mM |
| All canonical amino acids (except for the one to replace) |
50 mg/L |
| Ca2+ | 1 µg/mL |
| Fe2+ | 1 µg/mL |
| Trace elements (Cu2+, Zn2+, Mn2+, MoOH2+) |
0.01 µg/mL |
| Thiamine | 10 µg/mL |
| Biotin | 10 µg/mL |
Table 1. Composition of NMM19 chemically defined bacterial growth medium after preparation according to step 2.
