General scheme of neural progenitor isolation, cultivation, H3K79me2 ChIP and ChIP analysis methods: Figure 1 shows a flowchart to perform H3K79me2 ChIP of cortical progenitor cells at different time points during embryonic brain development or of cerebellar granular neuron progenitors in postnatal stages. As a first step, the brain has to be isolated and the telencephalon (between E11.5 and E14.5) or the cerebellum (P5-P7) has to be retrieved. It is possible to analyze the different regions of the telencephalon by dividing it into the DT and VT. Afterwards the tissue will be homogenized and the neural progenitors segregated. Now, it is possible to culture the progenitor cells and treat them with DOT1L inhibitors. Another possibility is to fix the progenitors directly and subject them to the ChIP procedure. For ChIP, the chromatin will be sheared into fragments of 200-500 bp and incubated subsequently with magnetic-beads-coupled antibody against H3K79me2. After washing the beads and retrieving the DNA, the precipitated DNA can be analyzed by sequencing or by qPCR (Figure 1A). It is essential to have a good size distribution of the chromatin fragments after shearing, so it is advisable to check the fragment size with DNA gel electrophoresis or with other methods (examples in Figure 1B). When choosing genome-wide sequencing, ChIP-seq-reads for H3K79me2 occupancy will be provided as a fastq-file for further analysis (Figure 1C). The quality of the sequencing reads needs to be assessed by application of FastQC and, if necessary, the fastq-files can be trimmed using TrimGalore. Mapping to the Mus musculus genome mm9 or mm10 can be performed with Bowtie2 and controlled via PlotFingerprint. It is advisable to remove duplicates with MarkDuplicates and to normalize the reads by BamCoverage and to compare the ChIP to the input samples with BamCompare. The ChIP-seq reads can subsequently be visualized genome wide in heatmaps or gene-wise in Integrative Genomics Viewer (IGV) browser sessions.
CPC culture and DOT1L inhibition: CPCs were isolated and treated with 5 µM DOT1L inhibitor SGC0946 at day 0 and day 2. The solvent DMSO was used as a control. At day 3, CPCs were harvested, the proteins were isolated, quantified, and analyzed via immunoblot (Figure 2A). Figure 2A shows that H3K79me2 levels are effectively decreased after three days of CPC culturing and DOT1L inhibition ensuring an effective inhibitor treatment scheme. The protein amount of H3, GAPDH, or Tubulin-alpha serving as loading controls was thereby not impaired. Immunostaining of fixed CPCs with antibodies against active caspase 3 (CASP3+) for detecting apoptosis and HuC/D for visualizing neuronal cells indicated that treatment of CPCs with DOT1L inhibitor led to increased cell death after three days in culture, as shown in Figure 2B. Quantification of the cells which are CASP3+/DAPI+ or HuC/D+/DAPI+ within the CPC culture revealed a significant rise of CASP3+ cells revealing programmed cell death upon DOT1L inhibition. The number of HuC/D-positive neurons, however, remained unchanged (Figure 2C).
H3K79me2 ChIP-qPCR of Aft4, Ddit3, Scd1, Aft3, and Npm1 from CPCs treated with DOT1L inhibitor: To test if the SGC0946 inhibitor treatment of DOT1L was efficient and led to a reduction of H3K79me2 at specific genes, we performed a ChIP analysis followed by qPCR of CPC treated for 3 days. Rabbit IgG was used as a ChIP control. Since it is known that H3K79me2 is located at the endoplasmatic stress-responsive genes Aft4, Ddit3, Scd1, and Aft3 as well at the nuclear transport gene Npm133,38, we used qPCR primers covering the TSS, the TES, and genomic regions within the gene bodies to determine differences in H3K79me2 distribution after DOT1L inhibition (Figure 3A). Genes with high H3K79me2 levels in the first place such as Atf4, Ddit3, and Npm1 were most responsive to the inhibitor treatment so that three days of inhibition of DOT1L led to a significant decrease of H3K79me2. Genes with a lower H3K79me2 coverage, such as Atf3 and Scd1, showed no significant change in H3K79me2 levels.
Overview of H3K79me2 ChIP-seq analysis: Genome-wide analysis of the H3K79me2 levels in CPCs from E14.5, performed and analyzed according to the presented schemes and protocols (Figure 1), revealed a very high quality ChIP. Whereas the binned counts, sorted according to their frequency of the input reads in the Fingerprint blot (Figure 4A), were evenly distributed, the H3K79me2 counts showed enrichment at specific regions (bins ranked with 1), which displays successful accumulation of chromatin fragments modified at H3K79. A heatmap of H3K79me2 along genes between their TSS and their TES and +/-10Kb upstream or downstream shows (Figure 4B) that H3K79me2 peaks around the TSS and decreased towards the TES in general. There are genes, which are completely covered with H3K79me2, and genes, which have a peak only within the TSS region. Endoplasmatic-stress related genes such as Atf4, Ddit3, and the nucleophosmin Npm1 harbor, for instance, a high H3K79me2 level at the TSS, which is only slightly decreased along the whole gene body (Figure 4C). In contrast, Scd1 has a low H3K79me2 occupancy at the TSS and no H3K79me2 within the gene body. Atf3 as a last example has different sharp and low level H3K79me2 peaks along the gene body.
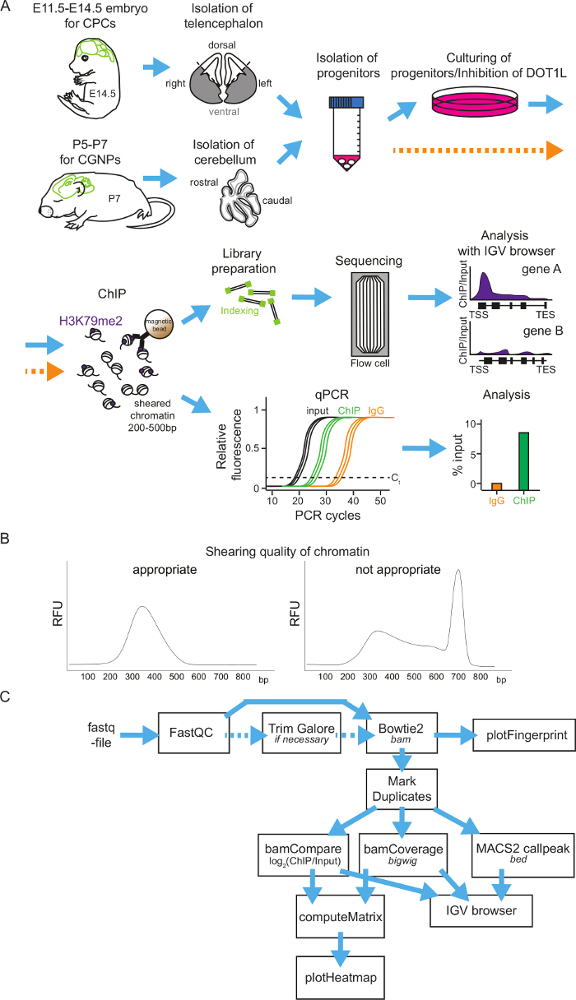
Figure 1: Scheme of H3K79me2 ChIP protocol from isolated CPCs or CGNPs and flowchart of sequencing analysis. (A) Overview of presented protocol. For CPC isolation, first E14.5 brains will be isolated and the cortices can be divided into DT and VT, if needed. For CGNPs, cerebelli have to be retrieved from P5-P7 mice. The tissue will be homogenized, the progenitors segregated, and then the cells can be cultured and treated with an appropriate inhibitor or directly used for ChIP. For ChIP, fixation of the chromatin is followed by shearing and immunoprecipitation with an anti-H3K79me2 antibody and rabbit IgG as control. After the DNA purification, ChIP samples can be analyzed via library preparation and sequencing or can be analyzed via qPCR. (B) Examples of appropriate and inappropriate quality chromatin. The DNA fragment distribution is shown in bp. (C) ChIP-seq results can be subjected to an analysis pipeline on the Freiburg/Galaxy server. After a quality control (FastQC), reads may be trimmed with TrimGalore and then mapped via Bowtie2 to the mouse genome (in the presented cases mm9). PlotFingerprint evaluates the quality of the ChIP. To define peaks for H3K79me2, MACSpeaks can be applied. For normalization BamCoverage and for comparison to the input BamCompare are usable. To visualize the results IGV browser and heatmaps are suitable. Expected file-formats are italicized. Please click here to view a larger version of this figure.
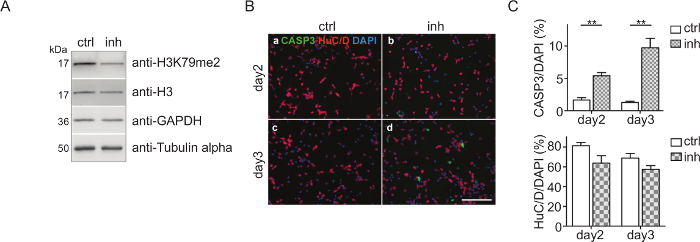
Figure 2: Cortical progenitor culture and DOT1L inhibition. (A) Immunoblots showing H3K79me2 levels in CPC at E14.5 after DOT1L inhibition for 3 days (day 3, n=3-11) in comparison to the DMSO control. H3, GAPDH, and Tubulin alpha were used as loading controls. (B) Immunocytological staining of a CPC culture at days 2 and 3. Activated Caspase 3 (CASP3 green)-positive cells and neurons (HuC/D: red) were stained. DAPI (blue) was used to visualize the cell nuclei. Scale bar: 100 µm. (C) Quantification of the immunostainings presented in (B). The ratio of positive cells for CASP3+/DAPI+ or HuC/D+/DAPI+ in percent is given. For statistical analysis, unpaired Student's t-tests were performed. p<0.01 **. This figure was modified from Roidl et al.33,38 Please click here to view a larger version of this figure.
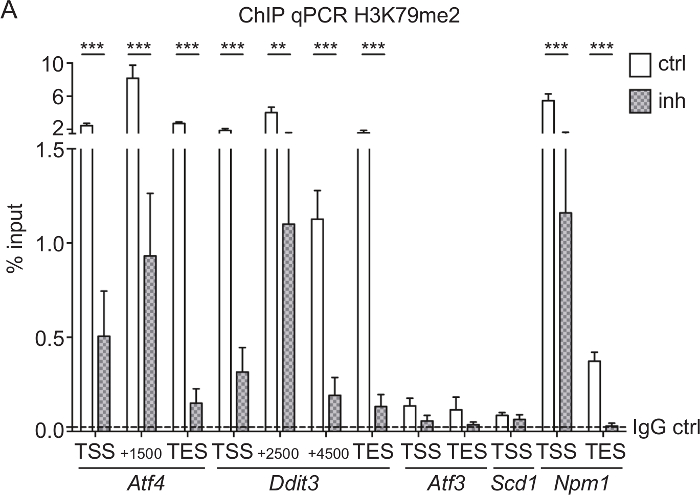
Figure 3: H3K79me2 ChIP-qPCR of Aft4, Ddit3, Aft3, Scd1, and Npm1 from CPCs treated with DOT1L inhibitor. (A) E14.5-derived CPCs were treated with 5 µM DOT1L inhibitor 4-5 h after isolation and for a second time at day 2 in culture. CPCs were fixed at day 3, which was followed by chromatin extraction, ChIP experiment, and qPCR. Results are represented as the mean (+/-SEM) % input (n=3). IgG was used as a negative control. The mean of the IgG level is depicted as dashed line. For statistical analysis, two-way ANOVA was applied. p<005 *; p<0.01 **, p<0.001 ***; TSS transcriptional start site, TES transcriptional end site. This figure was modified from Roidl et al.33,38 Please click here to view a larger version of this figure.
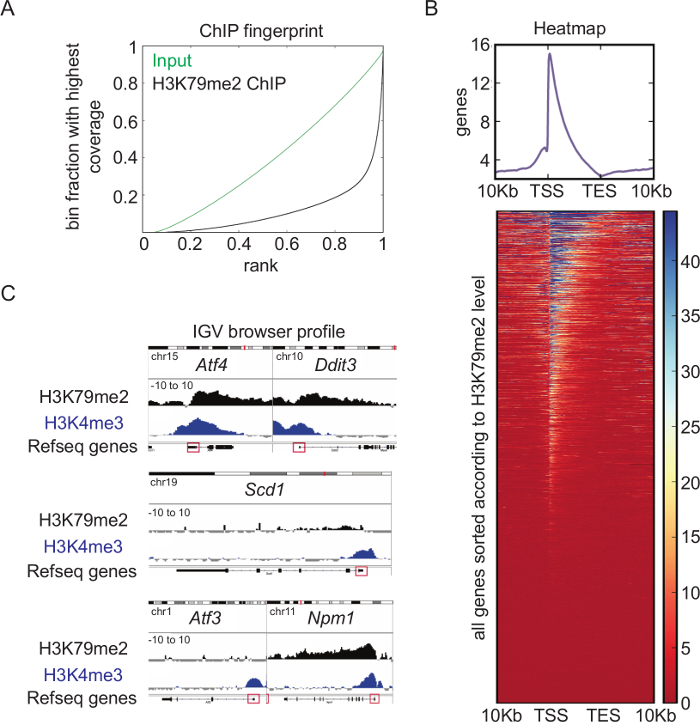
Figure 4: Overview of H3K79me2 ChIP-seq analysis. (A) Fingerprint of H3K79me2 ChIP-seq from E14.5-derived CPCs compared to input shows an enrichment of DNA fragments for H3K79me2 ensuring a high ChIP-seq quality. (B) H3K79me2 ChIP-seq results plotted in a heatmap. Strongly enriched genomic regions ARE presented in blue. Scaling 0 (dark red) – 45 (dark blue). H3K79me2 peaks near the TSS and declines in the 3´ direction to TES. (C) H3K79me2 ChIP-seq reads were mapped to the mm9 genome and visualized with the integrative genome viewer (IGV). The H3K79me2 occupancy of a gene is displayed as log2 of the number of reads ratio between ChIP and input reads per kilobase per million (Scaling: -10 to 10). Refseq gene structure is represented while a red box indicates the first exon including the TSS. The genes Aft4, Ddit3, Scd1, Aft3, and Npm1 are shown. For comparison, a H3K4me3 signal of the same genomic region is displayed. TSS transcriptional start site, TES transcriptional end site. This figure was modified from Roidl38. Please click here to view a larger version of this figure.
| Gene | Region | Primer forward 5'-3' | Primer reverse 5'-3' | ||
| Aft4 | TSS | GGACGATCTCTAACGCCACA | GCCCTAAACCCGCCCTTTAT | ||
| Aft4 | TSS+1500 | CTCCCGAATATGACATGAACCG | TCCATTCGAAACAGAGCATCG | ||
| Aft4 | TES | CCGTAATAGGGTAGTCAGGTGC | AAAGAATGACACTGAAAACCCACA | ||
| Ddit3 | TSS | GTACTGGCTCCGTCTAACCC | CAAGAGAGGGCCTGTAAGCA | ||
| Ddit3 | TSS+2500 | TCAAGCAGCCGGTCTCATAG | CTCAGATCCCCCAATGGCTT | ||
| Ddit3 | TSS+4500 | GAGCTGGAAGCCTGGTATGA | TCACCTCTTCGTTTCCTGGG | ||
| Ddit3 | TES | CACCAAGCATGAACAGTGGG | GTACCGTCTATGTGCAAGCC | ||
| Aft3 | TSS | AACTGAGAGCGCAACTCCTC | GCTGCCGTTCTTAGCTGGTA | ||
| Aft3 | TES | CTGTTGGCACAAAGTGGCTC | GGGATCTGCCATGGTGGAAA | ||
| Scd1 | TSS | TAGTGACCACACACAAAAGCTC | CCCAAGTGTAATTTGGATGATTTCC | ||
| Npm1 | TSS | TCCCCCTCCAGTCAGTTACC | CGTCCTTTCCTTGGCGTGA | ||
| Npm1 | TES | AGGGACATACTTAAGACAAGCCAG | AGGATTGAGGCAGACTGTCAAT | ||
| TSS:Transcriptional start site | |||||
| TES:Transcriptional end site | |||||
Table 1: List of primers used for ChIP-qPCR.
