In Utero Electroporation Approaches to Study the Excitability of Neuronal Subpopulations and Single-cell Connectivity
Özet
This manuscript provides protocols that use in utero electroporation (IUE) to describe the structural connectivity of neurons at the single-cell level and the excitability of fluorescently labeled neurons. Histology is used to characterize dendritic and axonal projections. Whole-cell recording in acute slices is used to investigate excitability.
Abstract
The nervous system is composed of an enormous range of distinct neuronal types. These neuronal subpopulations are characterized by, among other features, their distinct dendritic morphologies, their specific patterns of axonal connectivity, and their selective firing responses. The molecular and cellular mechanisms responsible for these aspects of differentiation during development are still poorly understood.
Here, we describe combined protocols for labeling and characterizing the structural connectivity and excitability of cortical neurons. Modification of the in utero electroporation (IUE) protocol allows the labeling of a sparse population of neurons. This, in turn, enables the identification and tracking of the dendrites and axons of individual neurons, the precise characterization of the laminar location of axonal projections, and morphometric analysis. IUE can also be used to investigate changes in the excitability of wild-type (WT) or genetically modified neurons by combining it with whole-cell recording from acute slices of electroporated brains. These two techniques contribute to a better understanding of the coupling of structural and functional connectivity and of the molecular mechanisms controlling neuronal diversity during development. These developmental processes have important implications on axonal wiring, the functional diversity of neurons, and the biology of cognitive disorders.
Introduction
The development of dendritic and axonal structures is an important facet of circuit regulation in the nervous system, including in the cerebral cortex. It plays a critical role during the selective wiring of the diverse neuronal subpopulations. A number of recent reports have shown that, in addition to connectivity, the molecular diversity of neurons is reflected by the acquisition of highly specific modes of firing. However, the mechanisms determining the excitability and connectivity of the distinct neuronal subtypes during development, as well as their degree of coordination, are still poorly understood1,2.
In vivo loss- and gain-of-function analyses allow for the study of the relationship between the expression level of specific genes and their influence in the development of the circuit. In utero electroporation (IUE) is a technique widely used to study the function of a gene of interest in specific neuronal populations and to study the overall patterns of their connectivity. However, to determine the morphological characteristics of axons and dendrites in cortical layers in living mice, it is essential to label neurons sparsely. A Cre recombination system combined with IUE can be used to mark a sparse population of neurons at a sufficiently low density to resolve the projections emitted by individual cells of the identified cortical laminas. This method labels a sufficient number of neurons per cortex to obtain quantitative data after the analysis of reasonable numbers of electroporated brains (Figure 1). This manuscript presents a method for such fine analysis of connectivity. It also presents a similar strategy to analyze, in separate experiments, the electrical properties of neurons by performing current-clamp recordings on green fluorescence protein (GFP)-electroporated cells from acute cortical slices. These protocols are versatile and can be applied to the study of the excitability and connectivity of neurons of WT and transgenic animals, and also of neurons in which losses and gains of function are introduced by additional plasmids during IUE.
Although this protocol describes the electroporation of mice at embryonic day (E)15.5, this technique can be performed at any age between E9.53 and postnatal day (P)24. While electroporation at early stages targets neurons and precursors of the thalamus and deep layers of the cortex, later-stage electroporation marks more superficial layers (e.g., E15.5 IUE targets layer II-III neurons). In summary, the combination of IUE with single-cell morphological analysis and electrophysiology is a useful tool to elucidate the molecular mechanisms underlying the enormous structural and functional diversity of neurons in the nervous system.
Protocol
All animal procedures were approved by the Community of Madrid Animal Care and Use Committee, in compliance with national and European legislation (PROEX 118/14; PROEX 331/15). Maintain sterile conditions during the procedure.
1. In Utero Electroporation
NOTE: This protocol for IUE is adapted from others that have been previously published5,6,7. This manuscript describes a protocol for the IUE of E15.5 embryos, with modifications in the reporter strategy that allow for the study of the morphology of single neurons8 and their electrophysiological properties in a separate experiment using standard GFP reporter plasmids.
- Preparing the DNA
- Prepare DNA plasmids using an endotoxin-free isolation kit as per the manufacturer's instructions and dilute them in 1x phosphate-buffered saline (PBS).
- For single-cell labeling, prepare 10 µL of DNA mixture per surgery (1 µL per embryo) using the following constructs and final concentrations: plasmid encoding a fluorescent protein reporter (e.g., CAG-DsRed29), 1 µg/µL as a control for electroporation efficiency; experimental plasmid to be tested, typically at 1 µg/µL; LoxP-stop-LoxP-fluorescent protein plasmid (CALNL-GFP10), 1 µg/µL; and construct-encoding Cre10, 1 – 4 ng/µL. Add 1 µL of 0.1% Fast Green in water (weight/volume) to visualize the injected DNA.
NOTE: Cre recombination of the GFP construct occurs only in a few neurons, which allows the visualization and reconstruction of individual axonal projections (Figure 1A). - For standard patch-clamp studies, prepare 10 µL of DNA mixture per surgery (1 µL per embryo) of the following plasmid encoding a fluorescent protein (e.g., CAG-GFP11), 1 µg/µL as a reporter control; experimental plasmid, generally 1 µg/µL; and 1 µL of 0.1% Fast Green in water (weight/volume).
NOTE: These are the standard electroporation conditions that analyze acute slices containing a large number of labeled excitatory neurons within the desired lamina. To perform patch-clamp studies in single cells, prepare the DNA as described in step 1.1.1.1.
- For single-cell labeling, prepare 10 µL of DNA mixture per surgery (1 µL per embryo) using the following constructs and final concentrations: plasmid encoding a fluorescent protein reporter (e.g., CAG-DsRed29), 1 µg/µL as a control for electroporation efficiency; experimental plasmid to be tested, typically at 1 µg/µL; LoxP-stop-LoxP-fluorescent protein plasmid (CALNL-GFP10), 1 µg/µL; and construct-encoding Cre10, 1 – 4 ng/µL. Add 1 µL of 0.1% Fast Green in water (weight/volume) to visualize the injected DNA.
- Prepare DNA plasmids using an endotoxin-free isolation kit as per the manufacturer's instructions and dilute them in 1x phosphate-buffered saline (PBS).
2. Preparation for Surgery
- Perform a survival surgery by using aseptic procedures. Ensure sterile conditions, including masks, gloves, instruments, and surgical field. Sterilize surgical instruments (scalpel, Adson forceps, hardened fine scissors, curved scissors, Dumont forceps, and needle holder).
- Select borosilicate glass capillaries of 1/0.58-mm outer/inner diameter. Pull capillaries3. Target for an optimal tip length of 1 cm after pulling. Cut the needle tip at approximately a 30° angle using fine forceps (Figure 1B).
- Prepare 500 mL of sterile isotonic solution (1x PBS or Hank´s Balanced Salt Solution (HBSS)). Add penicillin-streptomycin 1:100 and warm this solution to 37 °C. It can be stored at 4 °C after the surgery.
- Subcutaneously inject a preoperative dose of analgesics (e.g., carprofen, 5 mg/kg bodyweight).
- Keep the animals warm for surgery by placing them on a heating pad. Warm a clean cage to 37 °C for postoperative recovery.
3. Surgery
- Anesthetize a C57BL/6 E15.5 pregnant mouse with isoflurane. First, infuse a closed chamber with 3% isoflurane at 0.8 L/min oxygen and leave the mouse inside until it is asleep. Transfer the mouse to a heating pad and place the nose and mouth within a mask for delivery of isoflurane. Gradually decrease the anesthesia over the course of the surgery to 1.5% isoflurane via mask. Confirm proper anesthetization by observing a loss of the pedal reflex (toe pinch). The optimal procedure takes approximately 20 min and not more than 45 min.
- Apply eye ointment to prevent the eyes from drying during the procedure.
- Remove the hair from a ~ 3-cm region of the abdomen (using either an electric razor or depilatory cream). Wash the surgical area with cotton swabs infused with 70% ethanol, followed by an iodine-infused cotton swab. Repeat three times.
- Cover the surgical area with sterile gauze to prevent infections.
- Use the scalpel to make a vertical opening through the skin 2 cm long and parallel to the midline. Separate the skin and muscle of the abdomen using blunt curved scissors. Hold the muscle with the forceps and cut it through the linea alba to expose the abdominal cavity.
- Locate the embryos with the help of the forceps. Moisten two wet cotton swabs with the sterile saline solution prepared in step 2.3 and use them to manipulate an accessible embryo out of the opening. Place the cotton swabs around the embryo and gently extract both uterine horns from the abdominal cavity. Keep the embryos and the opened abdominal cavity hydrated with warm saline solution throughout the procedure to prevent them from drying out.
4. Injection of DNA and Electroporation
- Manipulate the embryos gently with fingers to locate the telencephalon (can be clearly visualized by the eye as the two more anterior vesicles of the brain). Place a prepared borosilicate capillary in a mouth pipette. Pass the tip of the needle through the uterus, avoiding blood vessels, until it reaches one lateral ventricle. Slowly inject approximately 1 µL of Fast Green-colored DNA solution until a large blue spot is observed.
- Place the 7-mm platinum electrodes laterally on the head of the embryo (Figure 1C).
NOTE: DNA is negatively charged; therefore, it moves toward the positive electrode when voltage is applied between the platinum electrodes. By varying the location of the electrodes, different areas of the brain can be targeted. - Apply voltage via the platinum electrodes (for E15.5 embryos: 5 pulses, 38 V, 50-ms interval cycle length, 950-ms interval pause. These conditions vary depending on the developmental stage)12.
NOTE: See Table 1 for voltage conditions and electrodes for different embryo stages. - Repeat steps 4.1 – 4.3 for each embryo.
5. End of the Surgery and Postoperation
- Use cotton swabs to manipulate the uterus back into the mother. Fill the abdominal cavity with the warmed saline solution (add about 2 mL).
- Suture the muscle with simple interrupted stitches or a continuous stitch. Use #6-0 sutures.
- Use staples to close the external wound. Be careful to separate the skin from the muscle before stapling. Remove the nose mask.
- Allow the mouse to recover for 30 min in the heated, clean cage before placing it in the animal facility room. Do not leave an animal unattended until it has regained sufficient consciousness to maintain sternal recumbency. Do not place an animal that has undergone surgery in the company of other animals until fully recovered.
- Supervise the animal during the days following the surgery. Apply analgesics subcutaneously (carprofen, 5 mg/kg bodyweight) every 12 h for 2 days or according to animal legislation. No additional postoperative care is required for the pups.
6. Preparation and Analysis of the Samples
- Optional: On P2, check for the expression of fluorescence in the electroporated pups using a fluorescent microscope; when the IUE is successful, fluorescence can clearly be seen in the head13. Mark or separate the mice that are positively electroporated for use in the following steps. Keep the dam and pups in standard animal facility conditions.
- At the desired stage of study, perfuse the mice transcardially. Typically, the most active phase of dendritic and axonal growth corresponds to the first three postnatal weeks2,14,15.
- Prepare 30 mL of 4% paraformaldehyde (PFA) in 1x PBS per mouse and chill it on ice.
CAUTION: PFA is a known allergen and carcinogen. It is toxic. Wear appropriate personal protective equipment.
NOTE: The amount of PFA needed depends on the age of the mouse (e.g., for P16, 20 mL for the perfusion and 10 mL for the postfixation are needed). - Prepare a ketamine-xylazine mix to anesthetize the animal with a 1:1:8 volumetric ratio of 100 µg/mL ketamine:100 µg/mL xylazine:PBS (prepare around 0.2 mL per mouse).
- Anesthetize the mouse by intraperitoneally injecting 0.2 mL of the mix prepared in step 6.2.2.
- Place the anesthetized mouse on its back. Make a horizontal incision above the thorax using a scalpel. Cut the muscle and the diaphragm with fine scissors until the heart can be observed.
- Make an incision in the right atrium with the fine scissors. Slowly inject 20 mL of PBS in the left ventricle with a 25-gauge needle in order to remove the blood. Inject 20 mL PFA (step 6.2.1) until the mouse is rigid and the organs become white.
- Immediately remove the head and make a midline incision in the skin from the neck to the skull. Gently peel away the skull using curved forceps and extract the brain. Take special care not to puncture or damage the brain during the removal of the skull. Put the brain in 10 mL of 4% PFA at 4 °C overnight to postfix it.
- Prepare 30 mL of 4% paraformaldehyde (PFA) in 1x PBS per mouse and chill it on ice.
- Cryoprotect fixed brains in 10 mL of 30% sucrose in PBS at 4 °C for 1 – 2 days, until they sink. Prepare 1-cm3 aluminum foil cubes. Fill the cubes 2/3 of the way with OCT. Put the brains inside and freeze them by putting the cubes on dry ice. Store the frozen brains at -80 °C.
- Place drop of OCT on the surface of the specimen disc, peel the aluminum foil from the histology cube, and position the cube at the desired orientation to the specimen disk on top of the liquid OCT. Apply firm pressure until it is fixed. Insert the specimen disc into the specimen head of the cryostat. Orient the specimen (move it into a favorable position relative to the knife/blade).
- Section the brains on the cryostat. Select 50 – 100 µm-thick floating cryosections16 using a fine brush and place them in PBS.
- Stain if desired.
NOTE: For the study of axonal morphology, staining with a GFP antibody is strongly recommended2.- Block the sections for 1 h at room temperature with 5% fetal bovine serum in PBS containing 0.5% Triton-X 100 (blocking solution). Incubate overnight at 4 °C with 1:500 primary antibody (e.g., rabbit anti-GFP) diluted in blocking solution.
- Wash the sections three times in PBS. Add 1:500 secondary antibody (e.g., goat anti-rabbit Alexa Fluor 488) diluted in blocking solution and incubate for 1 h at room temperature. Wash the sections three times in PBS.
- Counterstain with DAPI (1:10,000) in PBS containing 0.5% Triton-X 100 for 10 min. Rinse the sections with PBS. Mount the sections in an aqueous mounting medium16.
7. Imaging and Analysis
- Fluorescence or confocal microscopy
- To reconstruct complete neurons, acquire the images with a high magnification (at least 40X) and high resolution (minimum 1,024 x 1,024). Select the "Tile scan" or equivalent option in the acquisition software to cover the area of interest, spanning all dendrites and axonal processes. Acquire a sufficient number of stacks on the z-axis to avoid a loss of information.
NOTE: Generally, the microscope's software has an "Optimized stack" option, but if this is not the case, test manually to determine how many steps are necessary for the images to overlap in order to correctly define individual projections. Two crucial parameters are the pinhole and the objective; take into account "higher magnification, more resolution, and more necessary z-steps."
- To reconstruct complete neurons, acquire the images with a high magnification (at least 40X) and high resolution (minimum 1,024 x 1,024). Select the "Tile scan" or equivalent option in the acquisition software to cover the area of interest, spanning all dendrites and axonal processes. Acquire a sufficient number of stacks on the z-axis to avoid a loss of information.
- Analysis
NOTE: Although several parameters can be analyzed, step 7.2.1 focuses on two: dendrite morphology and axon branching. Download Fiji (http://fiji.sc/).- Open the image, select the "Segmented line" option from the menu. Draw a line (moving up and down along the z-axis by scrolling the mouse) following the structure of the neuron. Go to "Analyze, Tools, ROI Manager, Add" (alternatively, press "t") to save the line. Repeat this process for every axon or dendrite of the analyzed neuron2.
- In the ROI Manager Menu press the "Measure" button to get the length (or additional parameters). Export the measurements to a text file or spreadsheet to analyze them.
8. Electrophysiology
NOTE: The goal of this protocol is to obtain whole-cell current-clamp recordings from layer II/III pyramidal cell neurons identified visually by GFP expression in GFP-electroporated mouse brains (or any other fluorescent protein previously electroporated). It is an adaptation of previously published methods17,18. Using this protocol it is possible to study the effect of a genetic modification introduced by IUE on the electric properties of the neuron. The acquisition of specific firing modes is a gradual process of differentiation that involves the dynamic expression of a wide repertoire of ion channels and that results in the expression of transient firing modes before late postnatal stages. For example, mature electrical responses are not observed in layer II/III of the somatosensory mouse cortex before P162,19.
- Prerequisites for the acute slices
- Prepare sterile surgery tools to remove the brains from the mice: a guillotine, to remove the head; small scissors, to cut through the skull; forceps, to separate the skull from tissue; a spatula, to delicately remove brain tissue from its casing; a metallic slicer, to dissect the cortex into two equal halves; and a Pasteur pipette, to move slices from the vibratome (place them into a solution containing artificial cerebrospinal fluid (ACSF) for inspection, and then transfer the slices from ACSF to an incubation holding area).
- Prepare 1 L of ACSF using water of high purity (double-distilled water) containing 119 mM NaCl, 26 mM NaHCO3, 11 mM glucose, 2.5 mM KCl, 1.2 mM MgCl2, 2.5 mM CaCl2, and 1 mM NaH2PO4. Titrate the pH to 7.3 – 7.4 with HCl or NaOH. Adjust the osmolarity to 290 mOsm.
- Bubble ACSF with carbogen (95% O2/5% CO2) for 15 – 20 min using Teflon tubes (~ 1 mm), to stabilize the pH to 7.3 – 7.4.
- Freeze 200 mL of ACSF solution saturated with carbogen at -80 °C for 10 – 15 min to generate a slushy dissection solution. Place a 100- x 20-mm tissue culture dish on ice for slicing the brain.
- Prepare 100 mL of 1% low-melting point agarose solution just before the start of the experiment. Cool the solution, cut out a square piece of agarose (dimensions: 1 x 1 x 0.5 mm), and superglue it to the back of the platform, right behind where the brain is sliced (agarose gel provides support for the brain during slicing). Make the front as flat as possible.
- Obtaining acute slices
- Immobilize the mouse and anesthetize it with isoflurane 2%. Place the head into the guillotine opening and decapitate it swiftly. Remove its skull as fast as possible with the use of bone rangers or fine forceps. Put the brain into chilled ACSF.
NOTE: It is important to perform this step quickly. - Place the brain in the chilled culture dish. Cut off the cerebellum with small scissors.
- Pick up the brain with a spatula and blot it dry on a paper towel.
- Glue the ventrocaudal plane of the brain on a vibratome holder. Place the holder on a vibratome filled with ice-cold ACSF.
- Obtain acute slices by cutting 300-µm coronal sections (ensure continued carbogenation throughout the procedure, e.g., place a bubbler (1-mm polytetrafluoroethylene tube) in the vibratome chamber) using the following vibratome settings: 0.06-mm amplitude and 0.08 – 0.10-mm/s speed. Set the advance to the slowest possible speed (~ 22 s per pass). Modify these optimal settings and obtain optimal conditions for each machine empirically if necessary.
- Incubate the acute slices for at least 60 min in ACSF supplemented with 3 mM myo-inositol, 0.4 mM ascorbic acid, and 2 mM sodium pyruvate while bubbling with 95% O2/5% CO2 gas. Maintain the temperature at 25 °C.
- Perform the slicing procedure in less than 15 min. If desired, the slices can be stored for 1 – 7 h before being transferred to the recording chamber for use.
- Immobilize the mouse and anesthetize it with isoflurane 2%. Place the head into the guillotine opening and decapitate it swiftly. Remove its skull as fast as possible with the use of bone rangers or fine forceps. Put the brain into chilled ACSF.
- Prerequisites for the whole-cell recording
- Make sure that the electrophysiology station is equipped with a recording chamber, a perfusion system, a microscope, electrodes (recording, stimulating, and ground), macro- and micromanipulators, a rigid vibration-resistant table-top and Faraday cage, a stimulator, an amplifier and analog-to-digital (A/D) converter, a computer with acquisition software, and a GFP (or any other fluorochrome) filter for analyzing genetically modified neurons18 (Figure 2A).
- Prepare the intracellular solution, containing 115 mM potassium gluconate, 2 mM MgCl2, 10 mM HEPES, 20 mM KCl, 4 mM Na2ATP, and 0.3 mM Na3GTP, adjusted to pH 7.2 by KOH and to 290 mOsm by KCl.
- Make patch pipettes by pulling borosilicate glass capillaries. Prepare patch electrodes using a micropipette puller. Use borosilicate capillaries (1.5-mm outer diameter, 0.86-mm inner diameter, 10-cm length). Make patch electrodes showing resistances of 3-10 MΩ when filled with intracellular solution.
- Perfuse the recording chamber with ACSF at a rate of 2 mL/min. Maintain the temperature of the chamber at approximately 33 °C.
- Whole-cell recording
- Transfer the slices into the recording chamber using a Pasteur pipette (cut off the long tip) or a small brush. Hold down the slice with a harp. Perfuse the slices constantly with ACSF at a rate of 2 mL/min.
- Patch a GFP-positive neuron.
- Put the slice into the recording chamber and find the area of interest through the microscope at low magnification (10X). Then, find a GFP-positive cell to patch using the 60x objective.
- Fill the recording electrode with intracellular solution. Use the syringe linked to the filter (4-mm filter) and micro-loader tip to fill the recording electrode with intracellular solution.
- Place the glass pipette in the pipette holder. Place the pipette tip in the bath and focus on the tip. Once the pipette is in the bath, apply positive pressure through the back pressure control system.
- Patch a neuron that is fluorescent (Figure 2B).
- Approach the cell of interest under visual guidance while maintaining back pressure in the pipette. Upon the appearance of a small dimple on the cell surface, release the pressure. At this point, a tight seal (resistance larger than 1 GΩ) may be formed. Otherwise, apply a light negative pressure (suction) to facilitate it.
- While the seal is being formed, bring the holding voltage clamp to -60 mV. Once the GΩ seal is formed, apply a pulse of suction to rupture the cell membrane beneath the pipette and go into whole-cell mode. See reference20 for more details.
- Record the activity using current-clamp conditions21. Once in whole-cell mode, switch from voltage-clamp to current-clamp mode and start recording. For example, to record cell excitability, apply 500 ms-long depolarizing current injections (100 – 400 pA).
- Calculate the firing rates by plotting the number of action potentials along the train for increasing input currents.
NOTE: Resting membrane potential, input resistance, and membrane capacitance may also be calculated from the recordings21.
- Calculate the firing rates by plotting the number of action potentials along the train for increasing input currents.
Representative Results
To characterize the morphological changes of neurons in detail and throughout development, it is essential to label neurons sparsely. A Cre-recombinase diluted system allows for the expression of a gene of interest in a sparse population of neurons, so that only those neurons that incorporate this enzyme express GFP (Figure 1A). Using this strategy, layer II-III is targeted and labeled by IUE at E15.5. CAG-DsRed2 at 1 µg/µL, is co-electroporated as a control and to identify positive electroporated brains in living animals. Importantly, after staining with anti-GFP antibody, the signal is strong enough to allow for the clear visualization of their dendritic morphologies and axons (Figure 1D and E).
After IUE and electrophysiology, the analysis of the parameters obtained from whole-cell recordings are used to compare the firing responses and excitability of electroporated cells under different conditions. Several parameters can be obtained. The parameters should be adapted to the particular study using specific patch-clamp analysis software. Figure 2C provides an example of the plot of the action potentials against the input current obtained from recordings of a WT layer II-III neuron that was electroporated at E15.5.
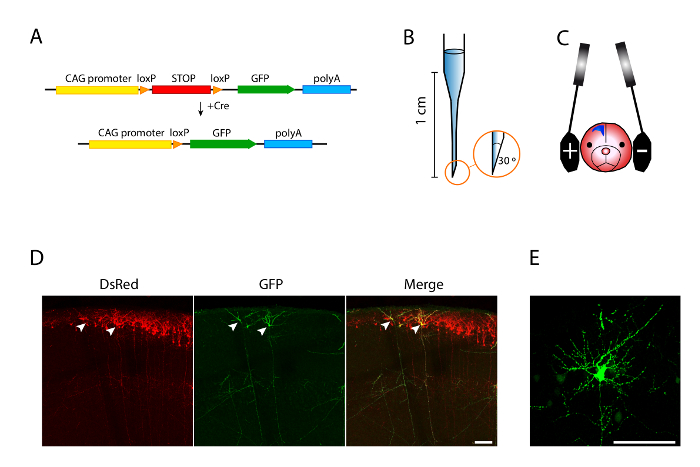
Figure 1. A Cre-recombinase Diluted Strategy Enables Sparse Labeling of Cortical Neurons. A. Schematic summary of the strategy. In neurons carrying both CALNL-GFP and CRE, the LoxP-STOP-LoxP cassette is excised out of CALNL-GFP, and GFP is expressed by the strong CAG promoter. B. Schematic drawing of a borosilicate capillary pulled using a micropipette puller. The tip is cut by forceps, creating a 30° angle. Measure 1 cm from the tip to the beginning of the narrower part of the capillary. C. Position of the electrodes to target the somatosensory cortex. The platinum electrodes are placed approximately over the ears of the embryo. Due to its negative charge, DNA goes toward the positive electrode when the voltage is applied. Variation in the position of the electrodes allows targeting different brain areas. D. Images obtained after vectors were delivered to layer II-III neurons by in utero electroporation at embryonic day 15.5; coronal sections were made at postnatal day 16. The CAG-DsRed2 vector was co-transfected as a control (left). GFP (middle) is expressed only in those neurons that also incorporated Cre, allowing the recombination of the LoxP sites in the CALNL-GFP vector. The sparse labeling allows individual neurons to be distinguished (arrowheads). E. High-magnification confocal image of the dendritic arbors of another sparsely labeled GFP neuron. Scale bars = 100 µm. Please click here to view a larger version of this figure.

Figure 2. Electrophysiology Settings and Example of a Firing Response. A. Photograph shows the electrophysiology setup used for patch clamp experiments in acute slices. The setup is included in a Faraday cage to eliminate noise, and the equipment is on top of an anti-vibration table. Controllers of the motorized micromanipulators for the electrodes are observed on the left. B. Pyramidal neuron of a mouse electroporated with GFP, observed under bright field and green fluorescence conditions. The recording pipette attached to a GFP+ cell is noticeable. Scale bar= 10 µm. C. Firing patterns of a CAG-GFP electroporated control layer II-III neuron showing the typical regular-spiking response. The distribution of action potentials approximates to a regular distribution along the duration of input current (X-axis). Please click here to view a larger version of this figure.
| Stage | Voltage | Electrodes | Referanslar |
| E9.5 | 7 V, 100 ms, 3 pulses | Stick platinum electrodes | Matsui et al., 20113 |
| E12.5 | 30 V, 50 ms, 3 – 5 pulses | Forceps-type electrodes 3 mm | Saito, T., 200612 |
| E15.5 | 35-48 V, 50 ms, 5 pulses | Forceps-type electrodes 5-7 mm | Rodriguez-Tornos et al., 20162, Saito, T., 200612 |
| P2 | 100 V, 50 ms, 5 pulses | Forceps-type electrodes 5-7 mm | Sonego et al., 20134 |
Table 1: Voltage Conditions and Electrodes for the Electroporation of Embryos.
Discussion
This protocol describes in detail how to label neurons of the somatosensory cortex of C75BL/6 mice in order to analyze their connectivity and their excitability. With respect to existing methods, it visualizes discriminating aspects of connectivity, such as the number of axonal branches per neuron, their precise topography, and their anatomical location. By altering the position of the electrodes, it is possible to target other neuron populations, such as the cingulate cortex (keep the same angle between the electrodes and the brain, but change the orientation of the poles) or the hippocampus5, and perform similar experiments labeling individual neurons or broader populations, depending on the desired strategy. However, there are limitations to this, as not all populations are equally accessible or equally selectively labeled. For example, in the hippocampus, it is possible to selectively target late-born neurons of the CA1 region, but early electroporation marks heterogeneous populations of inner and outer pyramidal cells. In the cerebral cortex, neurons are born in a sequential manner, so the gestation day during the IUE determines which cortical layer is affected. Performing earlier IUE targets deeper neurons (e.g., IUE at E14 labels layer IV neurons)22.
For a successful IUE, it is recommended to take into account certain considerations. First, it is important to do the surgery in less than 30 min in order to reduce the stress on the mother and to increase the chances of survival of the pups. Second, the most difficult part of the procedure is the injection of the DNA—perform the injection via the borosilicate capillaries as gently as possible. If the embryos are pressed too hard, they can be harmed. In terms of troubleshooting the death of the embryos during DNA injections, beveling the tip with a 30° angle can increase the efficacy of this process. If a beveller is not available and the capillaries are cut solely with forceps, the correct angle can be confirmed in the dissecting microscope. Discard inadequate capillaries. Finally, adapting the electroporation conditions to the stage of the embryo is important in order to increase the survival rate (see Table 1).
Some considerations are necessary with regard to the reconstruction of axons and dendrites. To label individual neurons, the proper concentration of the Cre plasmid are essential to obtain a good, sparse expression and to avoid the confounding overlap of neuronal projections belonging to different neurons. Although this protocol proposes the use of 4 ng/µL, it may be necessary to adjust the plasmid concentration for each experiment, depending on the promoter used, the quality of the DNA preparation, and the method of DNA quantification (e.g., reduce it to 2 ng/µL if labeling too many neurons). In addition, for axonal tracking, it is important to cut at an appropriate angle in order to have the whole neuron in the same plane.
Critical steps for successful patch-clamp recordings are the health of the tissue of the acute slices and the location and abundance of electroporated GFP-positive neurons. If patching steps fail or aberrant responses are found during the recordings, reduce the time for processing the acute slices. If GFP neurons are difficult to identify and locate due to their reduced numbers in the acute slice, ensure that sufficient CAG-GFP plasmid is included in the electroporation mix. With regard to the main limitations of the approaches described herein, the patch-clamp technique allows the recording of many different parameters describing the excitability of the neuron, but it does not evaluate aspects that depend on the whole circuit. Also, and as referred to above, not all neuronal subpopulations are accessible through IUE. In summary, in the future, these techniques can contribute to the further analysis of the structural and functional connectivity of different neuronal subpopulations in the brain.
Açıklamalar
The authors have nothing to disclose.
Acknowledgements
We are grateful to R. Gutiérrez and A. Morales for their excellent technical assistance and to L. A. Weiss for editing. C.G.B. is funded by the Spanish Ministerio de Ciencia e Innovación (MICINN), FPI-BES-2012-056011. This work was funded by a grant from BBVA Foundation and SAF2014-58598-JIN (MINECO) to M. Navarrete and by a grant from the Ramón Areces Foundation and grants SAF2014-52119-R and BFU2014-55738-REDT (from MINECO) to M. Nieto.
Materials
| pCAG-Cre | Addgene | 13775 |
| pCALNL-GFP | Addgene | 13770 |
| pCAG-DsRed2 | Addgene | 15777 |
| pCAG-GFP | Addgene | 11150 |
| Fast Green | Carl Roth | 301.1 |
| EndoFree Plasmid Maxi Kit | QIAGEN | 12362 |
| Carprofen (Rimadyl) | Pfizer GmbH | 1615 ESP |
| Isoflurane (IsoFlo) | Abbott (Esteve) | 1385 ESP |
| Ketamine (Imalgene) | Merial | 2528-ESP |
| Xylazine (Xilagesic) | Calier | 0682-ESP |
| Povidone Iodine | Meda | 694109.6 |
| Eye Ointment (Lipolac) | Angelini | 65.277 |
| Hanks' Balanced Salt Solution (HBSS) | Gibco by Life Technologies | 24020-091 |
| Penicillin-Streptomycin | Sigma -Aldrich | P4333 |
| Scalpel Handle #3 – 12cm | Fine Science Tools | 10003-12 |
| Scalpel Blades #10 | Fine Science Tools | 10010-00 |
| Adson Forceps-Serrated – Straight 12 cm | Fine Science Tools | 1106-12 |
| Hardened Fine Scissors – Straight 11 cm | Fine Science Tools | 14090-11 |
| Scissors Mezenbaum-Nelson Curved L=14,5cm | Teleflex | PO143281 |
| Thin curved tips – Style 7 Dumoxel | Dumont | 0303-7-PO |
| Dumont #5 Forceps-Inox | Fine Science Tools | 11251-20 |
| Mathieu Needle Holder – Serrated | Fine Science Tools | 12010-14 |
| AutoClip Applier | Braintree scientific, Inc | ACS APL |
| 9mm AutoClips | MikRon Precision, Inc. | 205016 |
| Sutures – Polysorb 6-0 | Covidien | UL-101 |
| Electric Razor | Panasonic | ER 240 |
| Borosilicate glass capillaries (100mm, 1.0/0.58 Outer/Inner diameter) | Wold Precision Instrument Inc. | 1B100F-4 |
| Aspirator tube assemblies for calibrated microcapillary pipettes | Sigma -Aldrich | A5177-5EA |
| Gauze (Aposan) | Laboratorios Indas, S.A.U. | C.N. 482232.8 |
| Cotton Swabs (Star Cott) | Albasa | – |
| Needle 25G (BD Microlance 3) | Becton, Dickinson and Company | 300600 |
| Sucrose | Sigma -Aldrich | S0389 |
| Paraformaldehyde | Sigma -Aldrich | 158127 |
| OCT Compound | Sakura | 4583 |
| Tissue Culture Dish 100 x 20 mm | Falcon | 353003 |
| GFP Tag Polyclonal Antibody | Thermo Fisher Scientific | A-11122 |
| Secondary Antibody, Alexa Fluor 488 conjugate | Thermo Fisher Scientific | A-11008 |
| DAPI | Sigma-Aldrich | D9542 |
| Fetal Bovine Serum | Thermo Fisher Scientific | 10270106 |
| Triton X-100 | Sigma-Aldrich | X100-500ML |
| Electroporator ECM 830 | BTX Harvard Apparatus | 45-0002 |
| Platinum electrodes 650P 7 mm | Nepagene | CUY650P7 |
| Microscope for Fluorescent Imaging – MZ10F | Leica | – |
| VIP 3000 Isofluorane Vaporizer | Matrx | – |
| TCS-SP5 Laser Scanning System | Leica | – |
| Axiovert 200 Microscope | Zeiss | – |
| Cryostat – CM 1950 | Leica | – |
| P-97 Micropette Puller | Sutter Instrument Company | P-97 |
| Patch clamp analysis softwarw (p-Clamp Clampfit 10.3) | Molecular Devices | – |
| Acquisition software (MultiClamp 700B Amplifier) | Molecular Devices | DD1440A |
| Motorized Micromanipulator + Rotating Base | Sutter Instrument | MP-225 |
| Air Table | Newport | – |
| Miniature Peristaltic Pumps | WPI | – |
Referanslar
- Dehorter, N., et al. Tuning of fast-spiking interneuron properties by an activity-dependent transcriptional switch. Science. 349 (6253), 1216-1220 (2015).
- Rodriguez-Tornos, F. M., et al. Cux1 Enables Interhemispheric Connections of Layer II/III Neurons by Regulating Kv1-Dependent Firing. Neuron. 89 (3), 494-506 (2016).
- Matsui, A., Yoshida, A. C., Kubota, M., Ogawa, M., Shimogori, T. Mouse in utero electroporation: controlled spatiotemporal gene transfection. J Vis Exp. (54), (2011).
- Sonego, M., Zhou, Y., Oudin, M. J., Doherty, P., Lalli, G. In vivo postnatal electroporation and time-lapse imaging of neuroblast migration in mouse acute brain slices. J Vis Exp. (81), (2013).
- Baumgart, J., Baumgart, N. Cortex-, Hippocampus-, Thalamus-, Hypothalamus-,Lateral Septal Nucleus- and Striatum-specific In Utero Electroporation in the C57BL/6 Mouse. J Vis Exp. (107), (2016).
- Petros, T. J., Rebsam, A., Mason, C. A. In utero and ex vivo electroporation for gene expression in mouse retinal ganglion cells. J Vis Exp. (31), (2009).
- Rice, H., Suth, S., Cavanaugh, W., Bai, J., Young-Pearse, T. L. In utero electroporation followed by primary neuronal culture for studying gene function in subset of cortical neurons. J Vis Exp. (44), (2010).
- Woodworth, M. B., et al. Ctip1 Regulates the Balance between Specification of Distinct Projection Neuron Subtypes in Deep Cortical Layers. Cell Rep. 15 (5), 999-1012 (2016).
- Wickersham, I. R., et al. Monosynaptic restriction of transsynaptic tracing from single, genetically targeted neurons. Neuron. 53 (5), 639-647 (2007).
- Matsuda, T., Cepko, C. L. Controlled expression of transgenes introduced by in vivo electroporation. Proc Natl Acad Sci U S A. 104 (3), 1027-1032 (2007).
- Matsuda, T., Cepko, C. L. Electroporation and RNA interference in the rodent retina in vivo and in vitro. Proc Natl Acad Sci U S A. 101 (1), 16-22 (2004).
- Saito, T. In vivo electroporation in the embryonic mouse central nervous system. Nat Protoc. 1 (3), 1552-1558 (2006).
- Bullmann, T., Arendt, T., Frey, U., Hanashima, C. A transportable, inexpensive electroporator for in utero electroporation. Dev Growth Differ. , (2015).
- Miller, M. Maturation of rat visual cortex. I. A quantitative study of Golgi-impregnated pyramidal neurons. J Neurocytol. 10 (5), 859-878 (1981).
- Miller, M., Peters, A. Maturation of rat visual cortex. II. A combined Golgi-electron microscope study of pyramidal neurons. J Comp Neurol. 203 (4), 555-573 (1981).
- Cubelos, B., et al. Cux-2 controls the proliferation of neuronal intermediate precursors of the cortical subventricular zone. Cereb Cortex. 18 (8), 1758-1770 (2008).
- Kang, J. Y., Kawaguchi, D., Wang, L. Optical Control of a Neuronal Protein Using a Genetically Encoded Unnatural Amino Acid in Neurons. J Vis Exp. (109), (2016).
- Mathis, D. M., Furman, J. L., Norris, C. M. Preparation of acute hippocampal slices from rats and transgenic mice for the study of synaptic alterations during aging and amyloid pathology. J Vis Exp. (49), (2011).
- Maravall, M., Stern, E. A., Svoboda, K. Development of intrinsic properties and excitability of layer 2/3 pyramidal neurons during a critical period for sensory maps in rat barrel cortex. J Neurophysiol. 92 (1), 144-156 (2004).
- Karadottir, R., Attwell, D. Combining patch-clamping of cells in brain slices with immunocytochemical labeling to define cell type and developmental stage. Nat Protoc. 1 (4), 1977-1986 (2006).
- Sakmann, B., Neher, E. Patch clamp techniques for studying ionic channels in excitable membranes. Annu Rev Physiol. 46, 455-472 (1984).
- Saito, T., Nakatsuji, N. Efficient gene transfer into the embryonic mouse brain using in vivo electroporation. Dev Biol. 240 (1), 237-246 (2001).
