To demonstrate the applicability of the BrUTP-strand-specific TRO procedure in detecting a transcription termination defect, we used a termination-defective, temperature-sensitive mutant of RNA14 called rna14-1. The role of Rna14 in termination of transcription by RNA polymerase II was demonstrated using the traditional TRO assay that detected RNA in the terminator-proximal region of selected genes1. The detection of RNA signal beyond the terminator region in the rna14-1 mutant at the non-permissive temperature was construed as the termination defect. The observed signal, however, could be ascribed to the pervasively transcribing polymerase that initiated transcription from somewhere near the 3′ end of the gene as shown in Figure 2B. To conclusively demonstrate the role of Rna14 in termination, we performed BrUTP-strand-specific TRO in the rna14-1 mutant and the isogenic wild type strain at 37 °C. We expected that in the wild type strain, the TRO signal will be constrained between the promoter and terminator regions as shown in Figure 1A. In the rna14-1 mutant, however, the polymerase will read through the termination signal and the TRO signal will be detected beyond the 3′ end of the gene as shown in Figure 1B and 2A.
Briefly, we grew the rna14-1 mutant and its isogenic wild type cells at 25 °C to mid-log phase and then shifted cells to 37 °C for three hours. Transcription run-on assay was performed to incorporate BrUTP into nascent RNA synthesized by an actively transcribing RNA polymerase as described above. The BrUTP labeled RNA was purified and reverse transcribed using primers C, D, E, F, and G shown in Figure 3A. The corresponding cDNA was PCR amplified using primer pairs B-C, B-D, B-E, B-F, and B-G and fractionated on agarose gels (Figure 3B). Quantification of gels is shown in Figure 3C. The presence of PCR amplified products from the primer pairs B-E, B-F, and B-G in the rna14-1 mutant reflects a termination read through phenotype (Figure 3B, Lane 11 – 13; and Figure 3C, regions E, F and G). In the wild type cells, however, the TRO signal was limited till the terminator element (Figure 3B, Lanes 2 and 3; and Figure 3C, regions C and D). There was no detectable TRO signal beyond the terminator region (Figure 3B, Lane 4 – 6; and Figure 3C, regions E, F and G). Thus, we could detect the promoter-initiated nascent transcripts that read through the termination signal in the mutant but not in the wild type cells, thereby confirming that Rna14 is a termination factor.
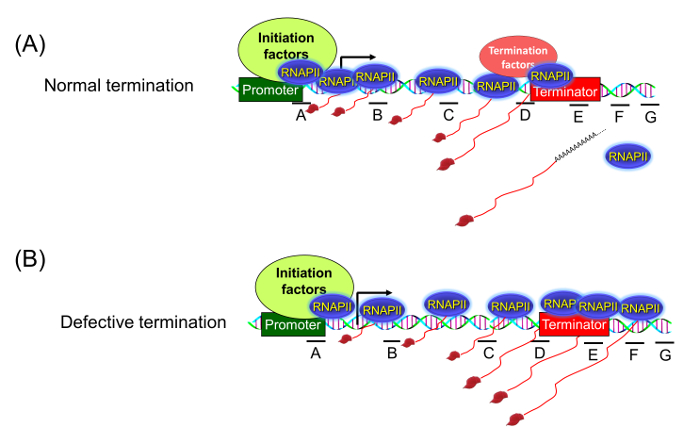
Figure 1: Transcription Run-on (TRO) Assay Detects the Presence of Transcriptionally Active RNA Polymerase in Different Regions of a Gene. (A) When termination is normal, there is no polymerase signal beyond the terminator region. (B) Upon defective termination, the polymerase is unable to read the termination signal and can be detected beyond the 3′ end of the gene. Please click here to view a larger version of this figure.
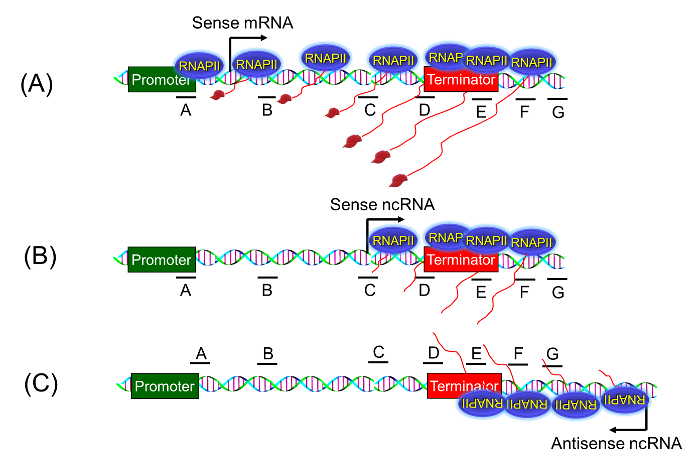
Figure 2: Strand-specific TRO Assay. The strand-specific TRO assay can detect if the presence of a transcriptionally active polymerase beyond the 3′ end of the gene is a true termination defect due to a promoter-initiated polymerase reading through the termination signal (A), or it is simply a pervasively transcribing polymerase transcribing in sense direction (B), or it is the anti-sense ncRNA transcribing polymerase initiating from the 3′ end (C). Please click here to view a larger version of this figure.
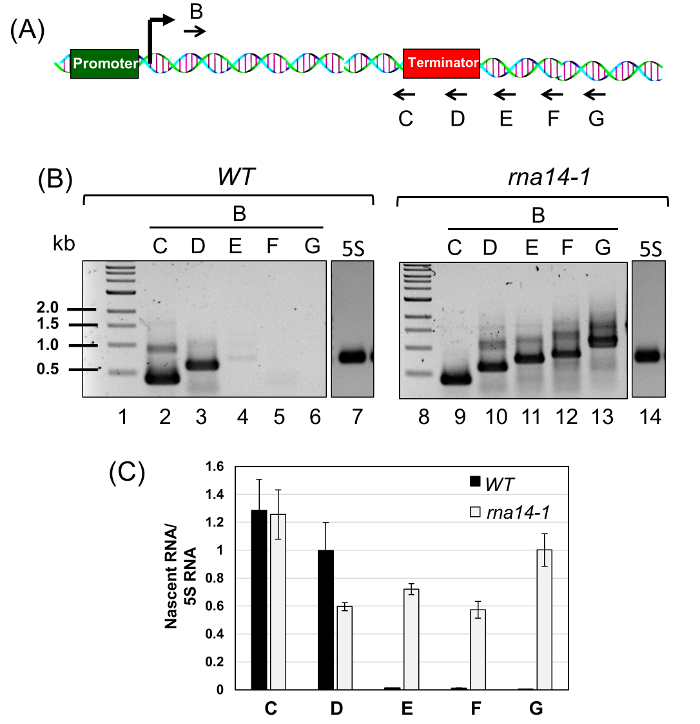
Figure 3: Rna14 is a Termination Factor as Polymerase Reads through the Termination Signal in the rna14-1 Mutant. (A) Schematic depiction of ASC1 gene showing the position of the forward primer B and five reverse primers, C, D, E, F, and G used for cDNA synthesis of purified BrUTP-labeled RNA. (B) cDNA made from reverse primers C, D, E, F and G was PCR amplified using the primers pairs B-C, B-D, B-E, B-F and B-G respectively. (C) The quantitative analysis of TRO data shown in (B) above in wild type and rna14-1 cells at 37 °C. 5S RNA was used as the normalization control. The error bars represent one full unit of standard deviation based on a minimum of three trials. Please click here to view a larger version of this figure.
