Identification of Alu-positive ADSCs in histological sections
Alu sequences are repetitive elements that comprise ~10% of the human genome and thus are excellent targets for identifying human cells in a species-specific manner43. In situ hybridization with DNA probes can be used to identify genomic elements on histological sections, including primary human cells29,30,40,44,45,46. Before examining the experimental sections, it is important to determine the morphology of the Alu-positive cells. Using a pellet of human cell spheroids (or of a cell suspension) as a positive control is highly recommended. It can be easily prepared by centrifuging and fixing a fraction of the prepared spheroids and processing them for paraffin sectioning, as described in step 3.1.8. Other chicken embryonic structures are used as a negative control. As can be observed, the nuclei of human ADSC spheroids (Figure 4F) or human grafted cells (Figure 4G) are Alu-positive, while chicken nuclei are Alu-negative (Figure 4G). The inability to find any human cells in the embryo is usually caused by the spheroid being dislodged after surgery. Carefully handling the chicken eggs after insertion of the spheroid, especially when sealing the egg with adhesive tape and returning it to the incubator, is important to avoid this issue.
Evaluation of the behavior of human ADSCs grafted into the somite region
As the method presented here is very flexible, the experimental design is crucial for meaningfully interpreting the behavior of human cells, as changes in the embryonic region or stage will expose grafted cells to a different microenvironment. ADSCs are a heterogeneous cell population obtained from adult white adipose tissue21,22,27,47. ADSC composition includes connective tissue, perivascular cells, and adipose progenitor/stem cells22. Thus, ADSC spheroids were grafted into the presomitic region at the wing bud level to investigate the potency and tropism of cells comprising this population. Presomitic mesoderm cells will form the somites, which can differentiate into diverse cell types, including bone/cartilage, muscle, dermis/adipose tissue, tendons, and perivascular cells48,49. In addition, neural crest cells migrate through this region and form diverse tissues, including melanocytes, dorsal root and sympathetic ganglia, peripheral nerves, and the adrenal primordia6. Thus, a spheroid grafted into the presomitic region will be exposed to signals that orchestrate the formation of multiple mesodermal and neural crest lineages.
Schematics summarizing the experimental workflow (Figure 1), as well as details of the preparation of spheroids (Figure 2), surgical procedures (Figure 3), and processing and hybridization with Alu probes (Figure 4) are shown. ADSC spheroids were transplanted into the 15th-20th presumptive somite region of embryos with at least 13 somite pairs (Figure 3J) at the wing bud level37. These 2-day-old embryos were reincubated until they were 3.5, 6.0, or 8.0 days old (Figure 5A). Some embryos were fixed only 4 h after the graft29 to easily identify Alu-positive human cells within the newly formed somite in the spheroid that had still not migrated (Figure 5B). Note the India ink deposit (asterisk) often found next to grafted cells (Figure 5C). Chicken cells are Alu-negative and smaller than human ADSCs (Figure 5C).
When embryos were incubated for a longer period, the human cells appeared more integrated into the chicken embryo29. At E3.5, Alu-positive cells were found distributed from the somitic region to the aorta-gonad-mesonephros region and dorsal mesentery, as well as perivascular to the dorsal aorta (Figure 5D,E). A fraction of cells were more ventral than the grafting site, indicating that a fraction of the ADSCs had migrated in the embryo. The HNK1 antibody (Human Natural Killer 1/CD57) can be successfully used in paraffin sections after in situ hybridization as it stains neural crest cells and nerve fibers50. Co-staining with HNK1 revealed that the human ADSCs had a tropism for neural crest cells and seemed to migrate alongside this tissue29 (Figure 5F).
Human cells could also be found when embryos were incubated until E6.029. As described in E3.5 embryos, cells were distributed from the mesenchyme lateral to the neural tube to the aorta-gonad-mesonephros region (Figure 5G,H). Some of the cells were perivascular to the dorsal aorta as well (Figure 5H). Co-staining with HNK1 revealed that many of these cells were associated with the peripheral nervous system, from the dorsal root and sympathetic ganglia to nerves extending until the aortic plexus (Figure 5I)29.
Categorizing the behavior of grafted cells according to location in the embryo
While additional experiments may help elucidate the fate of the xenograft in the chick embryo, the distribution of the Alu-positive cells does not require any additional experiments and is an important snapshot of possible cell migration from the graft region. We recommend evaluating the distribution to guide the following experimental steps before performing further experiments. Using a program such as Fiji's Cell Counter plugin is helpful for counting the cells.
Representative embryos were used to indicate regions of E3.5 and E6.0 embryos using a color code (Figure 5J and Figure 5L). In E3.5 embryos, most human ADSCs were found in the sclerotome (Figure 5K), a somite derivative43. Although obtained from lipoaspirate, most cells were not found in the dermomyotome, the tissue of origin for the chick dermis, or muscle cells44 (Figure 5K). Human cells were found in the mesenchyme lateral and ventral to the neural tube but were not located within the vertebrae cartilage, the back muscles, body wall, or the limb buds29. The similar distribution between E3.5 and E6.0 suggests that extensive migration of human ADSCs did not happen between these stages.
Additional experiments that may help to understand the behavior of grafted human cells
After identifying the location of the grafted human cells, co-staining may be performed as a general nuclear counterstain or to stain specific tissues. DNA in situ hybridization is an alkaline phosphatase-based assay that forms a purple-blue precipitate, which is often combined with brown, peroxidase-labeled immunostaining51. Such staining is found after co-staining with an HNK1 antibody in Figure 5F and Figure 5I above. Co-immunostaining can also be seen in Figure 6G,G' and Figure 7C–E. Additionally, Alcian blue stains the cartilaginous matrix, and its light blue precipitate is easily discernible from in situ hybridization (Figure 5I and Figure 6E,F,F'), although it stains the cartilage of older embryos more clearly (E8.0, Figure 7C,C').
Adjacent sections may be used to identify differentiation territories within the chick embryo or the tissues with which they are associated (such as nerves or vessels). Here, RNA in situ hybridization52 was performed to reveal Sox9-positive chondrogenic territories53 and Cbfa1-positive (or Runx2-positive) osteogenic territories54 (Figure 6A–C). Despite being found in the sclerotome of E3.5 embryos (Figure 5) and having skeletogenic potential in other models21,22,55, Alu-positive ADSCs did not co-localize with chondrogenic or osteogenic territories of E6.0 chick embryos29 (Figure 6A'–C'). Hematoxylin-eosin (HE) staining revealed that the human cells are at the border but not contained within the developing cartilage (Figure 6D) and show proximity to peripheral nerves (Figure 6D'). In older embryos, skeletogenic territories may be investigated using classical histological stains such as Safranin O, Masson's trichrome, or Chlorantine fast red/Alcian blue stains56.
Staining adjacent sections may also clarify whether the grafted cells are associated with any developing organ. A fraction of the ADSCs were found surrounding a rounded structure next to the dorsal aorta in E6.0 embryos29 (Figure 6E,F,F'). As this is the region in which the adrenal gland is formed, in situ hybridization with Bmp457 was performed to identify the presumptive cortical adrenal cells58. In addition, HNK1 immunostaining identifies the contribution of sympathoblasts that will form the adrenal medulla6. Staining an adjacent section with these markers (Figure 6G,G') revealed that some of the grafted ADSCs surrounded the adrenal primordia. Immunostaining with alpha smooth muscle actin (SMA), a marker for smooth muscle mural cells59, indicates that a fraction of the human cells is in a perivascular location (Figure 6H,H').
Interestingly, the xenograft affected chick morphogenesis in some embryos. In some E6.0 and E8.0 embryos, an ectopic structure can be found in the chick mesenchyme (Figure 7A and Figure 7C') (unpublished). This structure always appears innervated by HNK1-positive tissue (Figure 7B,C,C'). These data reinforce how human ADSCs may have paracrine effects on the environment. In the chick embryo, ADSCs had a strong tropism for neural crest-derived cells and tissues and had a positive effect on the growth of these structures.
Finally, it is possible to evaluate how many cells of the xenograft express a given marker or are associated with a given tissue. Co-staining with HNK1 allows the quantification of Alu-positive cells expressing HNK1 (black arrows), associated with chick HNK1-epxressing tissue but do not express HNK1 themselves (blue arrows), or neither of these situations (yellow arrows) (Figure 7D,E). This quantification reveals that most human ADSCs are associated with HNK1 in E6.0 embryos29 (Figure 7F). As a comparison, when human skin fibroblasts were grafted similarly, most cells did not express and were not associated with HNK1-positive tissue29(Figure 7F).
Evaluation of the behavior of human ADSCs grafted into the first pharyngeal arch
In light of the above results, it would be interesting to graft human ADSCs in a territory with a greater contribution of neural crest derivatives. The first pharyngeal arch has a contribution of neural crest cells that will form the skeleton and mesenchymal derivatives of the face6 and form melanocytes and the peripheral nervous system. This experiment would assay the chondrogenic and osteogenic potential of the ADSCs in the cephalic region, as these cells did not give rise to skeletal derivatives when grafted in the somitic region (Figure 5, Figure 6, and Figure 7) despite presenting this potential in vitro21,22 and in vivo in adult hosts55. Thus, a spheroid of human ADSCs was grafted in the region between the ectoderm and endoderm, lateral to the mesencephalon/first rhombomere, the presumptive first pharyngeal arch region38. The embryos were then reincubated until they were 4.5 days old (Figure 8A,B).
When the embryos were sectioned in the coronal orientation (Figure 8A), Alu-positive cells were found in the mandibular bud, a first pharyngeal arch derivative (Figure 8C,C'). Interestingly, these cells were associated with peripheral nerves, but not in regions that will form skeletal derivatives of the face (Figure 8D,D'). Thus, despite their in vitro and in vivo potential in adult hosts, ADSCs did not integrate into chondrogenic or osteogenic territories of the chick embryo's craniofacial region29. As shown earlier, the absence from skeletal territories was also observed when ADSCs were grafted at the wing bud level (Figure 5, Figure 6, and Figure 7). Interestingly, the ADSCs were associated with other neural crest-derived tissues, including the outflow tract (Figure 8E). This region of the heart receives an influx of cardiac neural crest cells, which form the arterial trunk60. A small fraction of the grafted ADSCs was found associated with the thyroid primordium29, another organ partially formed by neural crest cells6.
In conclusion, the human ADSCs had a tropism for neural crest cells in the trunk and cephalic regions, responding to signals that controlled both migration and morphogenesis of neural crest-derived tissues. The proportion of cells responding to these signals was higher than those reported for neural crest cell derivatives comprising the ADSC population28, suggesting that these responsive cells are not necessarily of a neural crest origin. These data may give insights into the possible role of peripheral nerves in regulating the behavior of cells contained within the ADSC population. Such regulation was described in detail in the bone marrow, where neural crest-derived mesenchymal cells are part of the hematopoietic niche61, and Schwann cells provide signals to promote quiescence of hematopoietic stem cells62. It was also reported that neural crest-derived cells from the endoneurium contributed to digit regeneration in mice63. Interestingly, it appears that the opposite was also true: ADSC also induced the growth of HNK1-positive nerves in the chick embryo (Figure 8I-L). These data reinforce the importance of investigating the role of peripheral nerves in the homeostasis of the stromal components of adult tissues.
Evaluation of the behavior of human primary glioblastoma grafted into the prosencephalon
The chick embryo xenograft model is also suited for the investigation of primary cultures of human tumors31. Primary human glioblastoma cells were grafted in the wall of the chick prosencephalon, which then developed until it reached HH24 or was 4 days old (Figure 8F,G). Transverse sections revealed that ~48 h after transplantation, the glioblastoma cell spheroid (or oncosphere) was not integrated within the chick neuroepithelium (Figure 8H,I). Interestingly, when embryos developed for 2 days, the glioblastoma cells disturbed normal prosencephalon development. A frontal view of the embryo reveals that the right telencephalic vesicle is reduced compared to the control side (Figure 8J). This reduction can also be observed in a histological section (Figure 8K), where a disruption in the continuity of the chick HNK1-positive neuroepithelium is identified (Figure 8L). The Alu-positive glioblastoma cells are found in the diencephalon (Figure 8L,M), where they are clearly not integrated into the neuroepithelium, as observed with HE staining (Figure 8N). In conclusion, the embryonic environment did not reprogram the glioblastoma cells, unlike what has been reported to take place when grafting human melanoma cells next to migratory neural crest cells15. However, glioblastoma cells survive within the chick embryo, making this xenograft model useful for testing the effects of chemotherapeutic drugs. We have previously used this model to reveal that the histone deacetylase inhibitor, trichostatin A (TSA), decreases the proliferation rate and downgrades the malignant phenotype of U87 human glioblastoma cells grafted into the chick prosencephalon31.
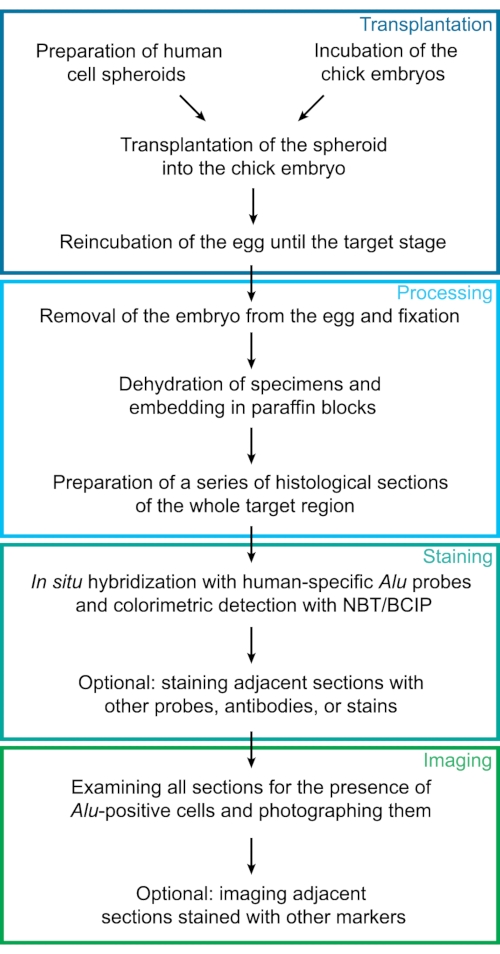
Figure 1: Experimental workflow. This diagram represents the experimental steps of the protocol described here. Individual steps were sorted into 4 major groups: transplantation, processing, staining, and imaging. Abbreviations: NBT = 4-Nitro blue tetrazolium chloride; BCIP = 5-bromo-4-chloro-3-indolyl phosphate p-toluidine. Please click here to view a larger version of this figure.
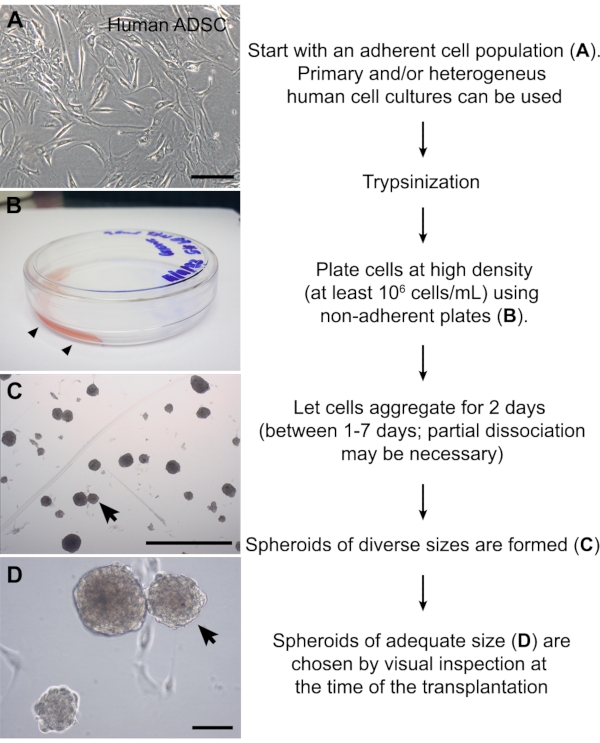
Figure 2: Preparation of cell spheroids. (A) Image of a human adipose-derived stromal cell culture at passage #6. (B) Non-adherent plate in which a small volume (1 mL) of cell suspension was carefully added to one side of the plate (arrowheads). The high cellular density promotes aggregation of the adherent cells. (C, D) ADSC spheroids 2 days after partial dissociation. This method yields spheroids of diverse sizes. A spheroid with the approximate size of the chick somite (~150 µm) is indicated (arrows). Scale bars = 100 µm (A, D) and 1 mm (C). Abbreviation: ADSC = adipose-derived stromal cell. Please click here to view a larger version of this figure.

Figure 3: Transplantation of the spheroid into the presomitic mesoderm of 2-day-old chick embryos. (A) Image of the workspace. Left side: stereomicroscope and gooseneck lamp. Right side: bottle of sterile PBS, India ink, dish with India ink solution in PBS, wide adhesive tape, a pair of microforceps, surgical scissors, slotted spoon, iris scissors, needle holder, egg holder, and Pasteur pipette. Surgical materials have been previously sterilized. (B) Thick capillary for transferring spheroids (left), thin capillary for injecting India ink (middle), and tip of the needle holder with an attached needle (right). (C) Zoomed-in image of the sharpened needle or microscalpel. (D) Illustration of a top view indicating the regions for cutting open the egg and for aspirating the albumen. (E) Illustration of a top view of an opened egg. If the egg has not been rotated in 24 h, the embryo will always sit on the top of the yolk. India ink should be injected into the yolk under the embryo. (F) Photograph of a 2-day-old chick embryo after injection of India ink. (G, H) A chick embryo with 14 somite pairs prior to the grafting of a spheroid, which had already been deposited next to the embryo (arrow). The last formed somite pair (14 s.) is indicated. A cut has been made (arrowhead) in the presomitic mesoderm of the presumptive 16th-17th somites into which the spheroid will be transplanted. (I, J) The same embryo after the transplantation is complete. Abbreviation: PBS = phosphate-buffered saline. Please click here to view a larger version of this figure.
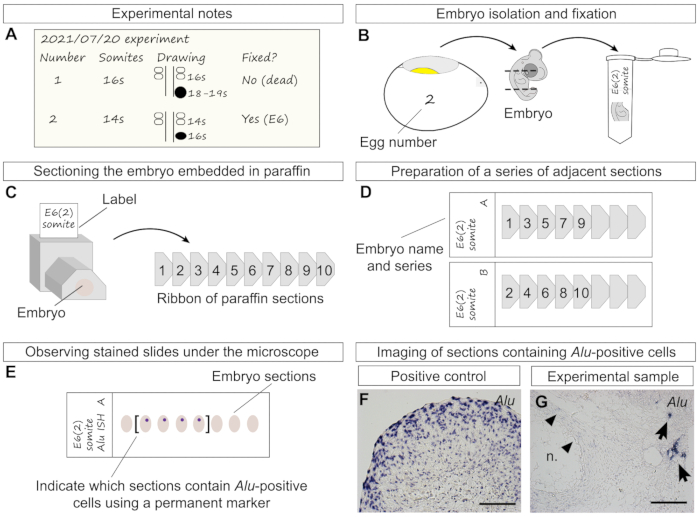
Figure 4: Additional experimental details from transplantation to staining. (A) Information that should be noted throughout the transplantation, including individual egg number, somite stage, and simple drawing of the spheroid position and size in relation to the two last formed somite pairs and neural tube. Later, the stage at which the embryo has been fixed should also be noted. Embryo "2" represents the transplantation performed in Figure 3G–J. (B) Representation of the removal of embryo "2" from the egg at age 6 days, decapitation, removal of the lower half of the trunk, and final transfer of the specimen to a tube containing fixative. (C) Left side: drawing of a trimmed and labeled paraffin block with embedded specimen in transverse orientation. Right side: a ribbon of sequential sections (right side) is obtained from cutting the paraffin block with the microtome. Numbers represent the order in which sections were cut, in an anterior-posterior order. (D) Example of slides prepared as a series of two adjacent sections (series "A" and "B"). The previously cut sections (C) were distributed between two slides, allowing the researcher to perform more than one type of staining of the same sample. (E) Human cells are stained purple-blue after in situ hybridization of Alu probes. Examining the slides and marking sections containing stained cells using a marker pen is recommended, especially sections of older embryos that may span several slides. (F) A large ADSC spheroid was processed for paraffin sectioning and in situ hybridization as positive control. All cells were stained with Alu probes. (G) In this experimental sample (6-day-old chick embryo), human cells were found next to the notochord after hybridization with Alu probes. Chicken cells were not stained. As light background staining may sometimes be found (arrowheads), it is recommended to use a counterstain. Scale bars = 100 µm (F, G). Abbreviation = n. = notochord. Please click here to view a larger version of this figure.
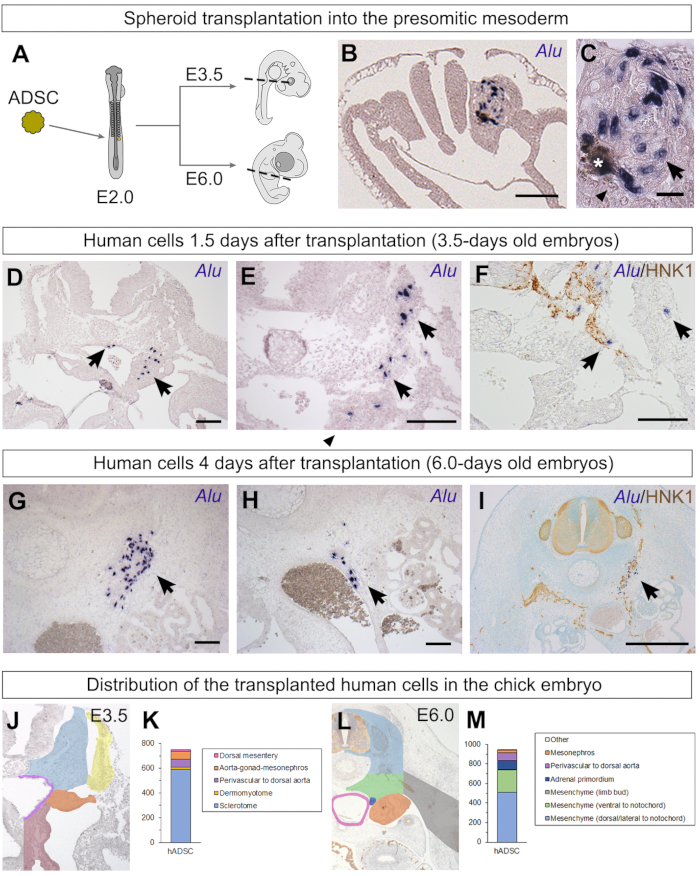
Figure 5: Identification of human ADSC spheroids grafted into the chick presomitic mesoderm and their behavior. (A) Illustration of the transplantation of an ADSC spheroid into the chick presomitic mesoderm. Stages in which embryos were fixed are also indicated, as well as sectioning planes (dashed lines). (B, C) When embryos were fixed only 4 h after the transplantation, the spheroid of human cells still had clearly defined borders29. Human nuclei were stained with Alu probes (arrows), while chicken nuclei were not stained (arrowheads). India ink inclusions were occasionally found next to the implantation site (asterisk). (D–F) When embryos were fixed 1.5 days after the transplantation, the human ADSCs (arrows) did not remain organized as a spheroid. Some cells were found lateral to the neural tube in the sclerotome, while others had migrated ventrally. Co-staining with Alu probes and HNK1, an antibody that recognizes neural crest cells and nerves, revealed a close association between some of the grafted human cells and migratory neural crest cells29. (G–I) When embryos were fixed 4 days after the transplantation, human ADSCs were scattered from the region lateral to the neural tube and notochord (G) to the region surrounding the dorsal aorta (H); many of the latter were perivascular cells. Co-staining with HNK1 revealed many human ADSC cells associated with the peripheral nervous system of the chick29 (I). (J–M) It is possible to classify the grafted human cells according to their distribution, revealing their possible migration from the graft site. Embryos from Cordeiro et al.29; graphs published here (K, 750 Alu-positive cells found in 4 E3.5 embryos; M, 945 Alu-positive cells found in 6 E6.0 embryos). Scale bars = 100 µm (B, D–H), 20 µm (C), and 500 µm (I). Abbreviations: hADSC = human adipose-derived stromal cell; HNK1 = Human Natural Killer 1/ CD57. Please click here to view a larger version of this figure.
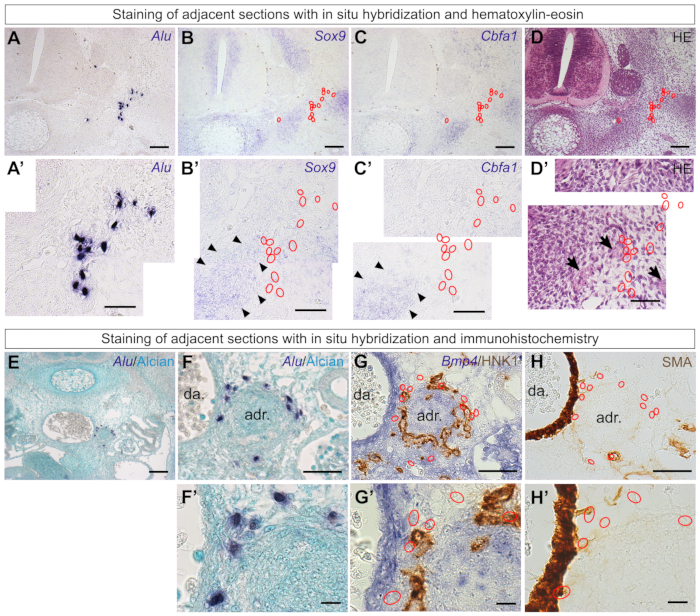
Figure 6: Staining adjacent sections of the same series clarifies the behavior of the grafted human cells. (A, A') Human ADSCs grafted into the chick embryo were located in an E6.0 embryo using Alu probes. (B–D) In situ hybridization of the adjacent sections with the chondrogenic marker Sox9 (B) or the osteogenic marker Cbfa1 (C), as well as observation of the tissue morphology with hematoxylin-eosin (D). (A'–D') Higher-magnification images of panels (A–D). Human cells (red outlines) were not located in skeletogenic territories of the chick vertebra (black arrowheads)29. Nerve fibers (black arrows) can be identified by their morphology with HE staining. (E, F) Human ADSCs were found in the aorta-gonad-mesonephros region of E6.0 embryos. (G). Staining of an adjacent section with Bmp4 probes and HNK1 antibody revealed that human cells (red outlines) were associated with the adrenal gland primordia of the chick. (H). Some cells had a perivascular location (smooth muscle actin)29. (F'–H') Higher-magnification images of panels (F–H). Scale bars = 100 µm (A–E), 50 µm (A'–D', F–H), and 10 µm (F'–H'). Abbreviations: ADSCs = adipose-derived stromal cells; HE = hematoxylin-eosin; da = dorsal aorta; adr = adrenal gland primordia; SMA = alpha smooth muscle actin ; HNK1 = Human Natural Killer 1/ CD57. Please click here to view a larger version of this figure.
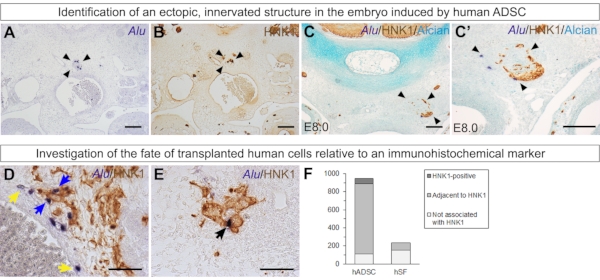
Figure 7: Additional experiments that can give insights into the behavior of grafted human cells. (A–C') In some embryos, an ectopic structure formed by human ADSCs (stained with Alu probes), chick mesenchyme, and chick HNK1-positive peripheral nerves was found (arrowheads). This supports the paracrine effect of ADSCs over their environment. (A, B) n = 2/6 E6 embryos. (C, C') n = 1/2 E8 embryo. (D–F) Quantification of grafted human ADSCs (945 cells, n = 6 embryos29) or skin fibroblasts in relation to HNK1-positive tissue in E6.0 embryos (235 cells, n = 3 embryos29). Alu-positive cells were classified as stained with HNK1 (black arrow), adjacent to HNK1-positive tissue (blue arrows), or neither stained nor adjacent to HNK1-positive tissue (yellow arrows). Scale bars = 100 µm (A–C, C') and 50 µm (D, E). Abbreviations: hADSC = human adipose-derived stromal cell; hSF = human skin fibroblast; HNK1 = Human Natural Killer 1/ CD57. Please click here to view a larger version of this figure.
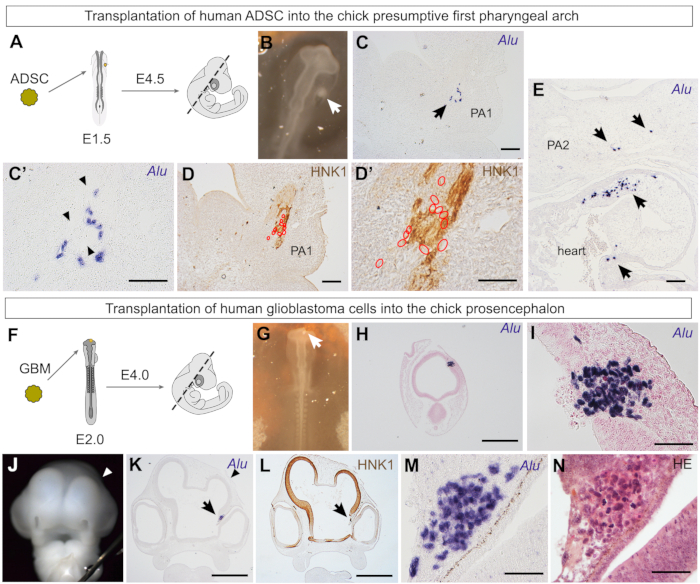
Figure 8: Investigation of the behavior of human ADSCs and glioblastoma cells grafted in the cephalic region of chick embryos. (A) Illustration of the transplantation of an ADSC spheroid into the chick presumptive first pharyngeal arch region, fixation stage, and sectioning plane (dashed line). (B) Human ADSC spheroid (white arrow) grafted between the ectoderm and endoderm, toward which neural crest cells that form the first pharyngeal arch will migrate. (C) Alu-positive cells (arrow) were found in the mandibular bud, a first pharyngeal arch derivative (PA1). (C') Higher magnification revealed that human cells were associated with a structure with scant nuclei (black arrowheads). (D) Human cells (red outlines) were associated with an HNK1-positive peripheral nerve. (D') As seen in a higher magnification image, most human cells (red outlines) are HNK1-negative. (E) Human ADSCs (arrows) were also found in the outflow tract, a region with contribution of cardiac neural crest cells in the chick heart. PA2, second pharyngeal arch. (F) Illustration of the transplantation of a glioblastoma spheroid into the chick prosencephalon, fixation stage, and sectioning plane (dashed line). (G) Photograph of a spheroid of primary human glioblastoma cells grafted into the chick prosencephalic wall. (H, I) Four hours after implantation, human glioblastoma cells were found embedded into the wall of the chick neural tube. Alu-positive cells did not appear integrated into the neuroepithelium at this stage. (J) Photograph of a 4-day-old chick embryo, 2 days after implantation of glioblastoma cells. The right telencephalic vesicle (arrowhead) was smaller than the left (control) side in all embryos (n = 4). (K) Grafted glioblastoma cells (arrow) were found in the diencephalon next to the eye; the reduced telencephalon was also evident in histological sections (arrowhead). (L) HNK1 staining revealed that the neuroepithelium formation was disturbed by the grafted glioblastoma cells. (M, N) Higher magnification of Alu-positive glioblastoma cells (M), clearly discernible from chick cells after hematoxylin-eosin staining (N). Scale bars = 100 µm (C–E), 500 µm (H, K, L), and 50 µm (C', D',I, M, N). Abbreviations: ADSC = adipose-derived stromal cells; GBM = glioblastoma; HNK1 = Human Natural Killer 1/ CD57; HE = hematoxylin-eosin. Please click here to view a larger version of this figure.
Table 1: DNA in situ hybridization solution recipes. Abbreviations: SSC = saline sodium citrate; DEPC = diethyl pyrocarbonate; MAB = maleic acid buffer; NTM = NaCl-Tris-MgCl2; 4-Nitro blue tetrazolium chloride; BCIP = 5-bromo-4-chloro-3-indolyl phosphate p-toluidine. Please click here to download this Table.
