Apoptosis, classified as type 1 programmed cell death1, ensures an equilibrium between cell proliferation and cell death2. Apoptosis is essential during human development, post-injury, and for the prevention of diseases such as cancer3. Intrinsic and extrinsic apoptotic cell death signaling pathways4 cause sequential biochemical and morphological intracellular changes2,5,6. Morphological apoptotic features can be identified by microscopy, and the biochemical perturbation can be analyzed by biochemical assays, including flow cytometry (FC)7.
Flow cytometric analysis to identify apoptosis and understand the associated intracellular mechanisms has burgeoned over the last two decades8. FC is a scientific methodology that analyzes cells in a fluid that passes through single or multi-channel lasers (Figure 1)9,10,11. The cells in the fluid are focused into a single file by the fluidics system of the flow cytometer using hydrodynamic focusing. As cells pass through the laser, light is scattered or emitted from the cells. The scattered light can be in the forward direction (forward scatter) or toward the side (side scatter) and provides information about the cell size, and cell granularity or internal structures, respectively.
In addition, fluorescent reagents, such as fluorescent dyes or antibodies labeled with fluorophores, detect specific surface or intracellular structures or molecules. When the laser excites the fluorophores, light is emitted at a specific wavelength. Detectors–commonly photomultiplier tubes–quantify the scattered and emitted light from cell samples. The detectors produce a quantifiable current that is proportional to the light scatter and fluorescence emission. The electronic output is converted into digital signals by computing software to identify cell populations based on cell size, cell granularity, and the relative cell fluorescence of fluorophore-labeled molecules9,12,13.

Figure 1: Schematic describing the technical operation and workflow of traditional flow cytometry. Cells are stained with fluorescent reagents and probed by a laser. The fluorescence signals generated are detected and converted into an electronic output, which is further digitized and analyzed by computer software and statistical programs. Abbreviations: FSC = forward scatter; SSC = side scatter. Please click here to view a larger version of this figure.
FC is used in both research and health diagnostics. The two goals of FC in necrobiology are the elucidation of molecular and functional properties of cell death and the discrimination of various modes of cell death14,15,16,17,18. FC applications include cell enumeration, sorting of cell populations, immunophenotyping, biomarker detection (e.g., apoptosis biomarkers), toxicity studies, and protein engineering12. In addition, FC is commonly applied to health diagnostics to assist with the diagnosis and monitoring of patients with hematological malignancies. Advances in instrumentation, fluorophore detection, and detector systems are expanding FC applications to include imaging cytometry, mass cytometry and spectral cytometry with broader research applications12.
The flow cytometric analysis of apoptosis provides advantages over traditional techniques used in assessing cell health. FC can analyze many single cells in a heterogenous sample rapidly and reproducibly to estimate apoptosis3,5. The ability of FC to provide quantitative information on cell phenotypes on an individual cell basis, and avoid bulk analysis, offers superior sensitivity to Western blotting, enzyme-linked immunosorbent assays (ELISAs), fluorometry, and spectrophotometry techniques utilized in the analysis of apoptosis8,19. Furthermore, the relative ease of FC analysis in contrast to the cumbersome and poorly reproducible manual steps of Western blots and ELISAs is advantageous. The reproducible, accurate, and high-throughput analysis of FC therefore is beneficial in cancer research20.
FC also permits the simultaneous analysis of cell cycle parameters for healthy and abnormal apoptotic cell populations21. As apoptosis is a dynamic process, different methods can produce variable results and are dependent on the time-point at which the cells are harvested22. The simultaneous quantitative assessment of multiple parameters of cell phenotype permits the detection of minor subpopulations with high accuracy, e.g., rare cell subsets with a low frequency of 0.01% can be detected23. Multi-parametric FC analysis is especially useful as apoptotic death occurs along a spectrum of early and late biochemical changes with cells at various points along the apoptotic continuum. For example, the use of dual staining using Annexin V and propidium iodide in FC analysis of apoptotic cells enables the categorization of early apoptotic cells, late apoptotic cells, and dead cells24. The accurate detection of apoptosis at multiple stages avoids misclassification and false-negative results. Thus, multiparametric analysis by FC improves the overall specificity of detecting cell phenotypes and avoids the misclassification of minor populations. Furthermore, cell sorting by FC allows the isolation of cell populations with high purity for subsequent analysis7.
The disadvantage of FC includes the use of cells in suspension, which can be challenging in tissue analysis, as disaggregation of tissue into cells may alter cellular function19. Moreover, the lack of standardization of FC instrument setup, data analysis, and assay reports may cause variation in results19, emphasizing the need for optimally training FC operators to perform, and analyze and report data. For example, the ability of FC to discriminate true apoptotic debris from apoptotic nuclei requires i) proper acquisition settings, ii) the use of calibration beads to identify a diploid DNA peak, and iii) negative and positive cell controls that are cell-specific3. Furthermore, multiparametric analysis is limited by the number of detectors, and optimal compensation needs to be performed to avoid non-specific results and spillover of fluorescent emission when using multiple fluorescent reagents25. Advances in instrument and fluorophore technology have improved parameter detection to 30 parameters12.
The identification of apoptotic cell death is not always simple7, and sensitive and specific biomarkers should be considered. The Nomenclature Committee on Cell Death (NCCD) recommends that more than one assay be used to study and quantify the process of apoptosis26. Microscopy analysis for classical apoptotic features26 is also recommended to confirm apoptosis and avoid false-positive results7. Four cardinal biochemical features that span early and late apoptotic events are (1) loss of cell membrane asymmetry; (2) dissipation mitochondrial membrane potential (ΔΨm); (3) caspase activation; and (4) DNA damage26.
During early apoptosis, phosphatidylserine is externalized to the outer cell membrane27 and can be detected by Annexin V fluorescently labeled with phycoerythrin27,28,29. Furthermore, dual staining with the fluorescent DNA-binding dye, 7-aminoactinomycin D (7-AAD), distinguishes live, late-apoptotic, and dead cells. Therefore, early-apoptotic cells stain positive for Annexin V and negative for 7-AAD, in contrast to late-apoptotic cells, that stain positive for both dyes24.
Intrinsic apoptotic signals induce dissipation of the mitochondrial membrane potential (ΔΨm). The disrupted ΔΨm causes the release of early pro-apoptotic proteins from the mitochondrial intermembrane space into the cytosol27,29,30. Change in ΔΨm can be assessed by dual staining with the positively charged, lipophilic dye, tetramethylrhodamine ethyl ester, TMRE, and 7-AAD. The TMRE dye accumulates within the inner membrane of intact mitochondria when the membrane potential is high. Depolarized mitochondria demonstrate decreased fluorescence. Live cells with polarized mitochondria (intact mitochondrial membrane) stain positive for TMRE and negative for 7-AAD. Dead cells with depolarized mitochondria stain negative for TMRE and positive for 7-AAD31.
The caspases are a family of intracellular proteases that, when activated, signal and execute apoptosis26,27. The terminal executioner caspases (3,6,7) effect late apoptosis29,32,33. Caspase-3 and -7 activities can be measured by a fluorescently labeled substrate, which, when cleaved, binds to DNA and emits a fluorescent signal. Furthermore, any compromise to cell membrane integrity can be assessed by staining with 7-AAD. Apoptotic cells stain positive for the DNA-binding dye but negative for 7-AAD. Late-apoptotic and dead cells stain positive for both dyes34.
Late apoptosis is characterized by DNA damage27,29,35, which can be evaluated by phosphorylated ataxiatelangiectasia mutated kinase (ATM) and histone H2A.X. Double-stranded DNA breaks (DSBs) cause phosphorylation of H2A.X. Fluorescently labeled antibodies against ATM and H2A.X can determine DNA damage. Negative detection of both ATM and H2A.X indicate no DNA damage, while detection of both dyes indicates the presence of double-strand breaks in the DNA36.
Actinomycin D is a potent inducer of apoptosis and acts by binding to DNA to block transcription and translation events37. This study aimed to assess biochemical apoptosis induced by actinomycin D in the SiHa cell line by analyzing early- and late-stage biomarkers of apoptosis. Four biochemical biomarkers of apoptosis assessed the sequential steps in the apoptotic cascade that included loss of cell membrane asymmetry, change in mitochondrial membrane potential, activation of terminal caspases, and DNA damage.
NOTE: This protocol describes steps in the cell preparation, cell enumeration, cell staining, and analysis of actinomycin D-treated SiHa cells using flow cytometric commercial assays measured and analyzed on a benchtop flow cytometer (Figure 2).
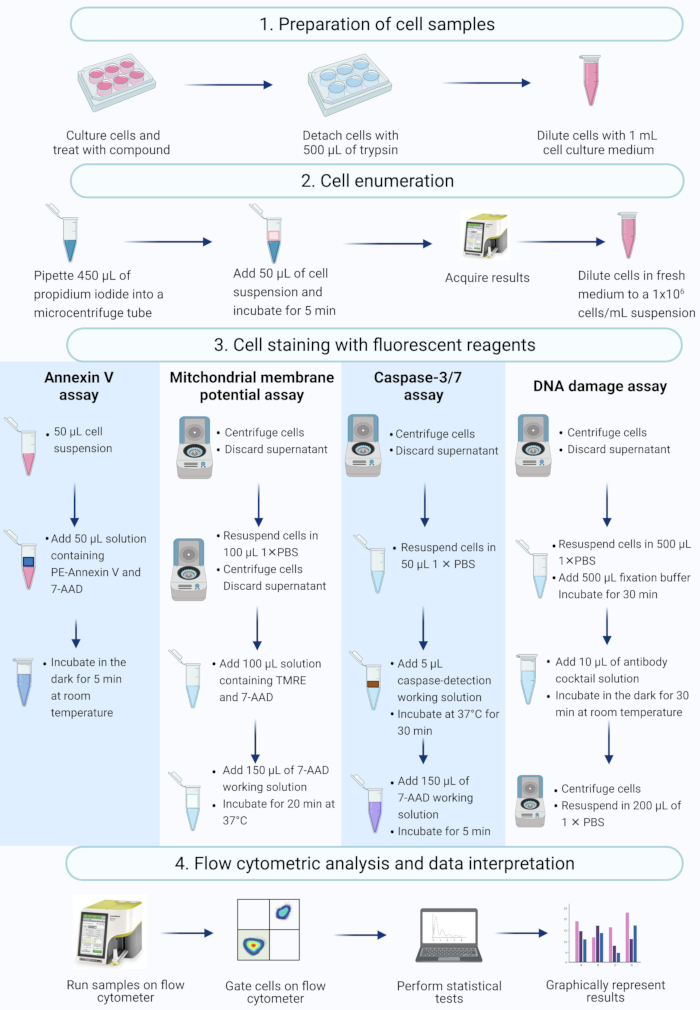
Figure 2: Workflow for the detection of biochemical apoptotic biomarkers by flow cytometry. Cells are cultured and treated as described in protocol step 1.1. (1) Cells are cultured, (2) enumerated and (3) stained with fluorescent reagents, and (4) analysed by a benchtop flow cytometer. Data are further gated, and statistically analyzed. Abbreviations: 7-AAD = 7-aminoactinomycin D; PBS = phosphate-buffered saline; PE = Phycoerythrin; TMRE = tetramethylrhodamine ethyl ester. Please click here to view a larger version of this figure.
1. Preparation of cell culture and treatments for flow cytometry
NOTE: Ensure aseptic technique is followed when handling cell cultures.
- Grow cell cultures in a humidified CO2 environment at 37 °C with 5% CO2. Ensure that cell culture is approximately 70% confluent in the parent flask before passaging cells for experiments.
- Remove the medium from the flask, wash cells with 1x phosphate-buffered saline (PBS) and add 500 µL of trypsin to detach the cells. After the cells begin to round and detach from the flask, sharply knock the base of the flask on the bench to detach the cells. Then neutralize the trypsin by adding approximately 5 mL of fresh culture medium supplemented with 10% fetal calf serum before counting the cells.
- Seed cells at 15,000 cells/mL in 3 mL of cell culture medium in a 6-well cell culture plate. Incubate the culture plates overnight at 37 °C with 5% CO2 to allow the attachment of the cells to the bottom of the well.
- Treat the experimental cultures with 100 ng/mL actinomycin D and the medium and solvent controls with fresh culture medium and dimethyl sulfoxide (DMSO), respectively, for 24 h. Collect the spent medium in a 15 mL tube and wash cells with 1x PBS. Thereafter, add the 1x PBS wash to the tube, and then add 500 µL of trypsin to the well. Next, neutralize the trypsin by adding 5 mL of fresh culture medium.
NOTE: (1) Prolonged trypsin incubation or incomplete neutralization may digest the cells and compromise their cell membrane, which may skew the results. In addition, it may also change cell membrane asymmetry and, thus, the accessibility to phosphatidylserine. (2) Cells that have detached and floated to the surface can be included in the subsequent analysis by centrifuging the removed medium at 300 × g for 5 min and resuspending the cells in fresh culture medium.
2. Enumerating cells using the viability assay
- Pipette 450 µL of propidium iodide in a microcentrifuge tube. Add 50 µL of the cell suspension to the microcentrifuge tube. Incubate the tube at room temperature for 5 min. Enumerate the cells by flow cytometry (refer to section 4).
- Dilute all samples to a concentration of 1 × 106 cells/mL with cell culture medium before proceeding to the apoptosis assays.
3. Staining cells for flow cytometry
NOTE: Sterile conditions are not required for this part of the protocol.
- Detection of phosphatidylserine externalization using Annexin V
- Add 100 µL of cell suspension into a microcentrifuge tube. Add 100 µL of a 1:1 mixture of fluorescent conjugated Annexin V and 7-AAD reagent. Incubate at room temperature for 20 min, protected from light.
- Analysis of mitochondrial membrane depolarization
- Centrifuge 100 µL of the cell suspension at 300 × g for 5 min and discard the supernatant.
- Resuspend the cells in 1 mL of 1x PBS, add 100 µL of TMRE staining solution to each sample, and mix the suspension by gentle back-pipetting.
- Incubate the cells for 20 min in a humidified CO2 environment at 37 °C. Wrap the samples in clean aluminum foil to protect from light.
NOTE: Mitochondrial membrane potential is a functional marker that is sensitive to minor changes to the cell environment. Therefore, samples should be incubated and measured under identical conditions (temperature, pH, and time elapsed between the onset of incubation and fluorescent measurement) to maintain reproducibility). Also, note that inadequate protection of samples from light causes photobleaching of fluorophores resulting in falsely low emitted fluorescence. - After incubation, add 5 μL of the 7-AAD staining solution to each sample and mix. Incubate for 5 min at room temperature, protected from light.
- Detection of activated terminal caspase-3 and -7 using caspase substrate DEVD
NOTE: The following solutions are prepared before performing this assay.- Dilute the DEVD-bound DNA-binding peptide in DMSO to a ratio of 1:8 with sterile 1x PBS to make the caspase-detection working solution. Store the solution on ice or at 2-8 °C, protected from light.
NOTE: Each sample will require 5 µL of this solution. - Add 2 µL of a 7-AAD stock solution to 148 µL of 1x PBS to make the 7-AAD working solution. Store the solution on ice or at 2-8 °C, protected from light.
NOTE: Each sample will require 150 µL of this solution. - Centrifuge 50 µL of the cell suspension for 5 min at 300 × g. Discard the supernatant. Resuspend the cells in 50 µL of 1x PBS, followed by 5 µL of the caspase-detection working solution. Mix thoroughly.
- Loosen the cap of the tubes and incubate for 30 min in a humidified CO2 environment at 37 °C, protected from light. Add 150 µL of the 7-AAD working solution to each sample and mix. Incubate for 5 min at room temperature, protected from light.
- Dilute the DEVD-bound DNA-binding peptide in DMSO to a ratio of 1:8 with sterile 1x PBS to make the caspase-detection working solution. Store the solution on ice or at 2-8 °C, protected from light.
- Detection of double-strand DNA breaks and total DNA damage
- Centrifuge 50 µL of the cell suspension for 5 min at 300 × g. Discard the supernatant.
- Resuspend the cells in 500 µL of 1x PBS. Add 500 µL of a formaldehyde-based fixation buffer and mix. Incubate the samples on ice for 10 min.
- Centrifuge for 5 min at 300 × g and discard the supernatant. Resuspend the cells in 90 µL of 1x PBS in a microcentrifuge tube. Add 10 µL of the antibody solution to the microcentrifuge tube. Incubate at room temperature for 30 min in the dark.
- Add 100 µL of 1x PBS and centrifuge for 5 min at 300 × g. Discard the supernatant. Resuspend the cells in 200 µL of 1x PBS.
4. Run samples on the flow cytometer.
- Verify the analytical performance of the flow cytometer by running the instrument's system check kit. Do not proceed with running samples until all checks are completed and passed.
- Locate the desired assay by browsing through the catalog of pre-programmed assays on the instrument and select Run Assay.
- Mix the sample by gently back-pipetting before loading the sample onto the flow cytometer.
NOTE: Adequate mixing ensures cells remain in suspension and prevents low cell counts. - First, load a negative control sample onto the flow cytometer, and select Run (Adjust Settings) so that the instrument begins aspirating the sample and provides a real-time preview of detected events. Refer to the Table of Materials for instrument name and details.
- Using the live preview, adjust the thresholds for fluorescence and cell size and draw a rectangular gate around the cell population. Drag the threshold marker to exclude cellular debris. Select Next (Set Health Profile) to proceed.
NOTE: It is important to know the size of the cells. Drag the sliders and observe changes in how detected events are plotted on the real-time preview, as this will inform an accurate selection of the thresholds. If cellular debris is not excluded at this stage, it cannot be removed in post-acquisition analyses. - Tap and drag the quadrant markers to separate the cell populations and so that the instrument plots detected events in real-time. Use these plots to guide the end-user on the appropriate placement of the quadrant markers. Select Next (Verify Samples) to proceed so that the instrument displays a summary of the settings. After reviewing the settings, select Next (Verify Settings) to apply these settings for all samples within the experiment.
NOTE: The instrument provides a 2-min live preview of cells used to adjust the instrument's settings. If this time limit expires, the instrument will release the sample, and steps 4.2.2-4.2.4 will have to be repeated. Remove the sample and mix well before reloading and continuing. - Gate a population of cells by drawing a region around the cell population. Adjust thresholds for fluorescence and cell size using the sliders located on the x- and y-axis of the live preview. Apply these settings for all samples within the experiment.
- Mix the first sample by gently back-pipetting and load the first sample onto the flow cytometer and select Next. Name the sample and select Run so that the system begins running the sample.
NOTE: The instrument can only run one sample at a time. - Once all samples have been run, save the experiment by giving it an appropriate title. Save the settings of the current experiment for retrieval in future runs (optional).
- Mix the sample by gently back-pipetting before loading the sample onto the flow cytometer.
5. Post-acquisition analysis
- If required, perform fine-tuning of gates or quadrant markers post-acquisition.
- Locate the experiment that needs adjustment by navigating the system's file browser and open the experiment.
- Tap on the thumbnail preview of the plot to enlarge it. Tap on the top-left or bottom-right corners of the cell gate to adjust the dimensions of the gate. To adjust the quadrant markers, tap on the intersection of the vertical and horizontal lines to move the markers as they are. To adjust the angle of either line, tap on the line and drag the handle.
- Adjust the markers (as previously described in steps 4.1.3-4.1.4) as desired and apply these settings to all samples in the experiment by selecting the check-mark icon, marking all the samples, and selecting Accept.
6. Statistical analysis
- Run tests in triplicate and run an analysis of variance (ANOVA) with a post-hoc Bonferroni test to assess significant differences between the treated samples and controls.
NOTE: The statistical test of choice is dependent on the investigator and the variables being analyzed.
The cell count and viability (Figure 3) results showed that 95.2% of the cells in the sample were live, and 4.8% were dead. The total cell concentration in the original sample was 6.20 x 106 cells/mL.
The Annexin V and cell death assay (Figure 4) showed a significant increase (p < 0.0001) in apoptotic cells in SiHa cells treated with 100 ng/mL actinomycin D in comparison to the controls. Because Annexin V staining is increased in cells during early stage apoptosis, this finding suggests that 100 ng/mL actinomycin D induced apoptosis in SiHa cells.
The mitochondrial electrochemical transmembrane potential assay (Figure 5) demonstrated a significant decrease (p < 0.0001) in cell health profiles (live, depolarized and live, depolarized, and dead, dead) between actinomycin D, medium control, and solvent control. These data suggest that 100 ng/mL actinomycin D induced mitochondrial depolarization in SiHa cells.
The caspase 3/7 assay (Figure 6) demonstrated significant (p < 0.0001) activation of caspases 3 and 7 in SiHa cells treated with 100 ng/mL actinomycin D compared to the controls. These finding demonstrates that 100 ng/mL actinomycin D induced apoptosis in SiHa cells.
The DNA damage assay (Figure 7) showed that 100 ng/mL actinomycin D significantly (p < 0.0001) induced DNA damage markers, ATM and H2A.X, in SiHa cells. This finding suggests significant increase in DNA damage in SiHa cells treated with actinomycin D.
The results of suboptimal experiments (Figure 8) demonstrate analytical considerations across all assays. Cell concentration affects accuracy of data. In Figure 8A, the cell count is unacceptably low. The gated cell populations in all 4 quadrants of the dot plot have low signal intensity. The manufacturer optimizes the assay for 300-700 cells/µL in the final sample volume. This example illustrates the importance of using the correct sample concentration prescribed by the manufacturer.
In addition, high cell concentrations also caused erroneous results (Figure 8B). Medium and experimental cultures demonstrated 99.95% and 100% live cells, respectively. The flow rate for both assays exceeded the manufacturer's optimized concentration of 100-500 cells/μL and required dilution with 1x Assay Buffer to avoid inaccurate analysis.
Cell clumping should be avoided during the preparation of experimental cultures as it produces false results due to increased cell size indices, as illustrated in the Annexin V assay. Figure 8C shows a dual problem of high cell concentration that exceeds the manufacturer's instruction and cell clumping, as evidenced by cell size indices exceeding 4 in the SiHa medium control. The high cell concentration is illustrated by the sheeting of cells forming a bright red plane of cells in medium cultures, demonstrating discordantly high apoptotic cell populations.
Prolonged staining of cultures can result in non-specific binding of proteins and result in false results, as demonstrated by the Annexin V assay. Figure 8D demonstrates similar results for medium and experimental cultures due to prolonged staining.
Poor sample handling, extended trypsinization of adherent cells, vigorous pipetting during wash steps, and high-speed and prolonged centrifugation steps cause cell lysis and high amounts of cell debris. In Figure 8E, cultures analyzed by the caspase 3/7 assay demonstrate increased cell debris evidenced by a small cell size index (<2.2 cell size index). Care should therefore be taken when preparing samples for data acquisition.
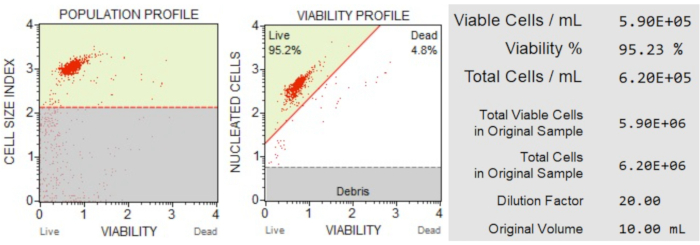
Figure 3: Cell Count and viability assay. The population profile separates debris from live and dead cells. The two-dimensional dot plot is gated and divides live and dead cell populations. The information panel provides quantitative data on cell count, percentage of total viable cells, and the total number of viable cells in the sample. These data can be used to standardize the cell count in all samples for subsequent apoptosis analyses. Please click here to view a larger version of this figure.
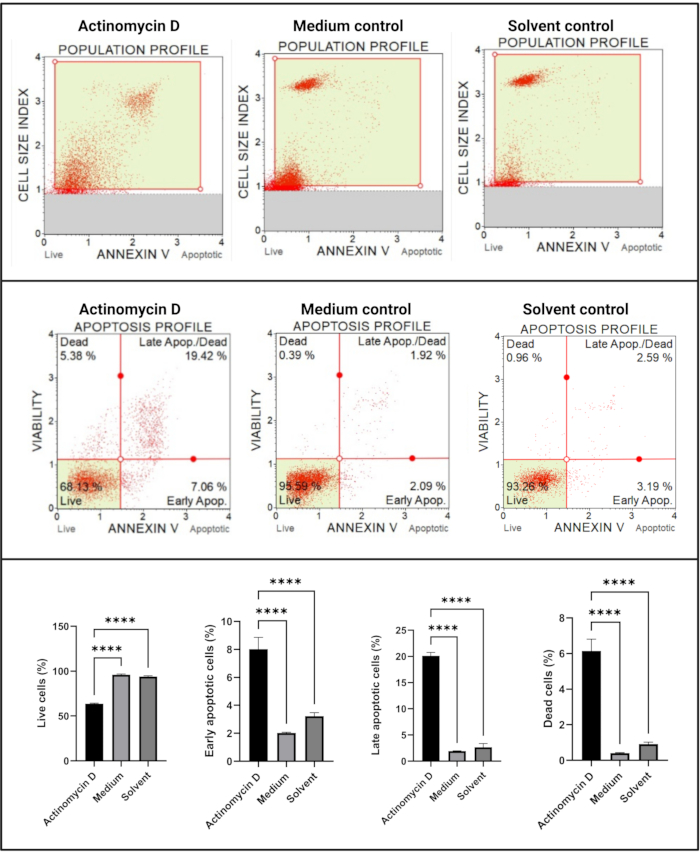
Figure 4: Annexin V assay. All samples show identical gating parameters, and statistical analysis of each sub-population reveals a significant increase in apoptosis in cells treated with actinomycin D. All assays were performed as three independent experiments, and each experiment was assayed in triplicate. Data are presented as mean ± SD, and p < 0.05 was considered statistically significant. **** p < 0.0001. Please click here to view a larger version of this figure.
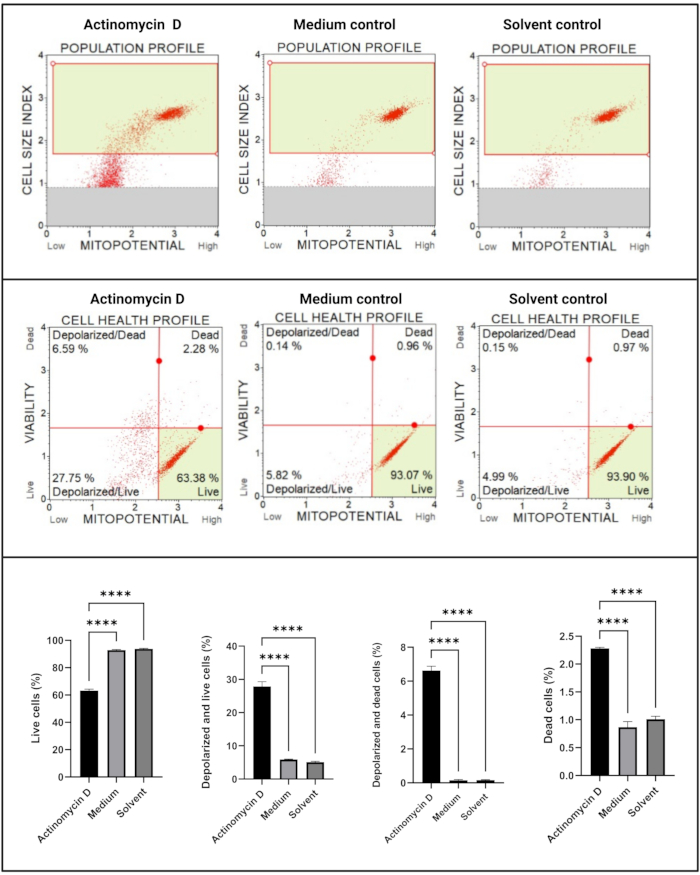
Figure 5: Mitochondrial electrochemical transmembrane potential assay. All samples show identical gating parameters, and statistical analysis of each sub-population reveals significant perturbation in mitochondrial membrane potential in cells treated with actinomycin D. All assays were performed as three independent experiments, and each experiment was assayed in triplicate. Data are presented as mean ± SD, and p < 0.05 was considered statistically significant. **** p < 0.0001. Please click here to view a larger version of this figure.
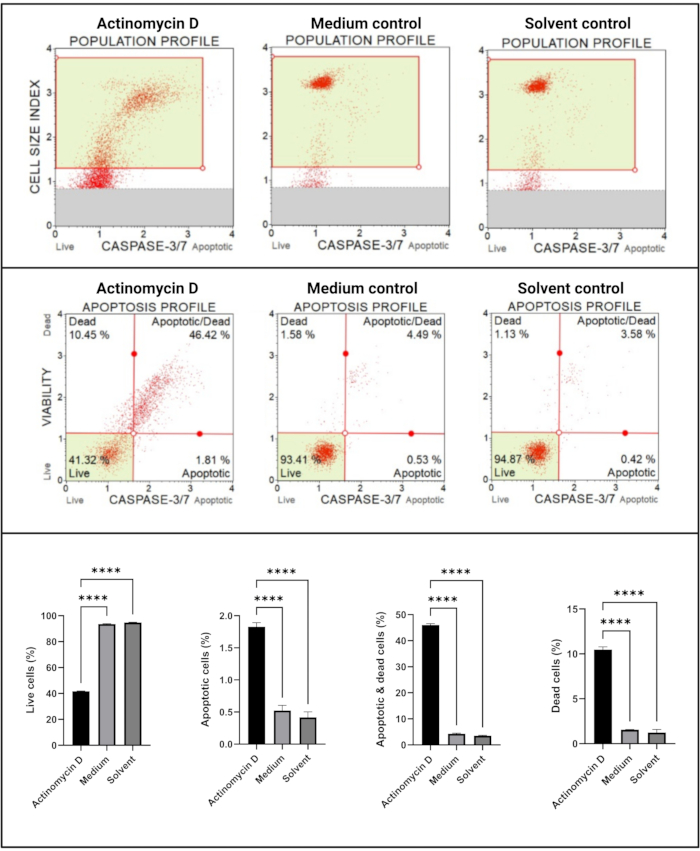
Figure 6: Caspase 3/7 detection assay. All samples show identical gating parameters, and statistical analysis of each sub-population reveals a significant increase in caspase 3/7 activity in cells treated with actinomycin D. All assays were performed as three independent experiments, and each experiment was assayed in triplicate. Data are presented as mean ± SD, and p < 0.05 was considered statistically significant. **** p < 0.0001. Please click here to view a larger version of this figure.
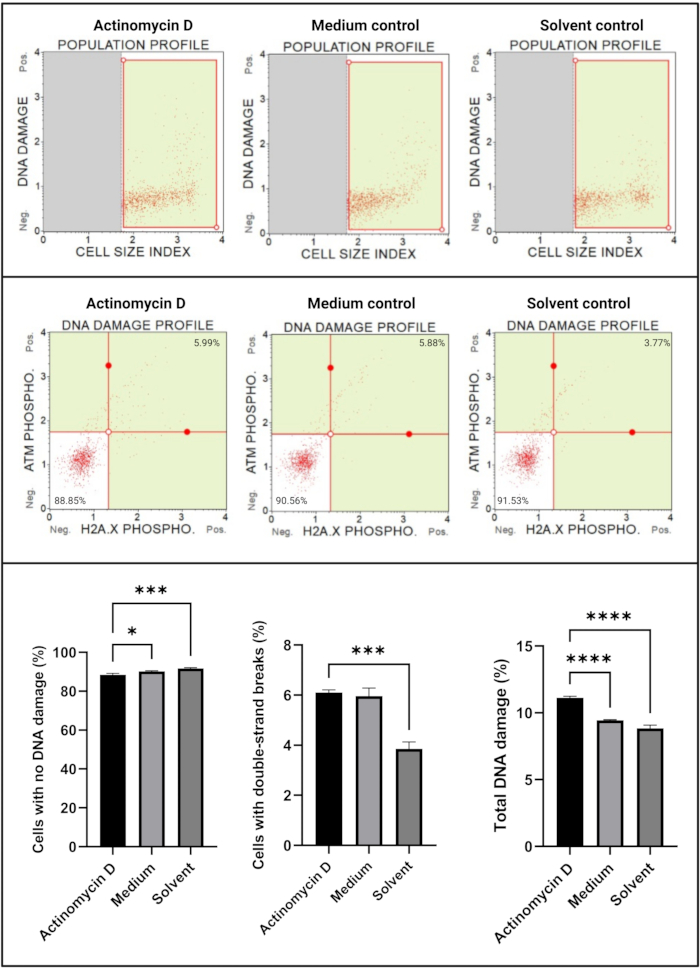
Figure 7: DNA damage assay. All samples show identical gating parameters, and statistical analysis of each sub-population reveals a significant increase in double-strand DNA damage and total DNA damage in cells treated with actinomycin D. All assays were performed on three independent experiments, and each experiment was assayed in triplicate. Data are presented as mean ± SD, and p < 0.05 was considered statistically significant. * p < 0.05; *** p < 0.001; **** p < 0.0001. Please click here to view a larger version of this figure.

Figure 8: Sub-optimal experimental conditions yield poor results. (A) Low concentration of cells, (B) high concentration of cells, (C) cell clumping and aggregation evident by high cell size index, (D) prolonged staining of cell samples evident by increased positive staining in both samples, and (E) poor sample handling evident by increased cellular debris (cell size index < 2.2). Please click here to view a larger version of this figure.
