All the experiments using mice and rats were approved and conducted using Schedule 1 procedures covered under the Home Office Animals (Scientific Procedures) act 1986. The artificial cerebrospinal fluid (ACSF) recipe can be found in Table 1.
1. Acute Hippocampal Slice Preparation
- Remove the mouse brain as quickly as possible, using a spatula and sharp scissors to cut the skull, and place it in ice-cold, oxygenated (95% O2, 5% CO2), high-sucrose ACSF (~100 mL).
- Place the mouse brain on a Petri dish and remove the cerebellum and a small section of the frontal cortex with a scalpel before hemisecting down the midline of the brain.
- Place the two hemispheres on the side that was just cut and glue each hemisphere onto the stage of a vibratome.
- Cut 200 – 300 µm-thick sections of the complete region of interest. In the case of the hippocampus, approximately 3 – 4 slices per hemisphere should be obtained.
- Using a plastic Pasteur pipette, transfer the slices to a chamber submerged in oxygenated (95% O2, 5% CO2) ACSF. Maintain at 34 °C for 30 min.
NOTE: The treatment of slices with active pharmacological agents, such as recombinant Dkk1 proteins, can be performed at this stage by adding the agents to the slice chamber. - Remove the slices from the chamber using a paintbrush, place them into a 24-well plate, and fix for 20 min to 1 h in 4% paraformaldehyde (PFA)/4% sucrose at room temperature (RT).
Caution: PFA is toxic, wear appropriate protection. - Wash the slices three times in 1X PBS (10 min each).
NOTE: The protocol can be paused here for 1 – 2 days, as long as the slices are kept at 4 °C in 1X PBS.
2. Immunofluorescence for Synaptic Markers
NOTE: The recipes of all buffers used can be found in Table 2.
- Replace the PBS with blocking/permeabilizing buffer (Table 2) in the slice wells and incubate at RT for 4 – 6 h.
- Dilute the polyclonal guinea pig antibody against vGlut1 at a 1:2,000 dilution in blocking/permeabilization buffer.
NOTE: vGlut1 is a marker for pre-synaptic excitatory terminal visualization. For post-synaptic excitatory synapse identification, use a polyclonal antibody against PSD-95 and dilute 1:500 in the same solution. Use a minimum volume of 300 µL in each well. To identify the anatomical location of the hippocampus, use an antibody that can also identify the neuronal structure, such as MAP2 or Tubulin. Work out the correct concentrations of all primary antibodies for the synaptic markers of choice (pre- and post-synaptic) and dilute in fresh blocking/permeabilizing buffer. - Incubate the slices in the primary antibody solution overnight (or for 1 – 2 days) at 4 °C. Use a shaking platform with vigorous movement.
- Wash the slices three times in PBS (10 min each).
- Dilute the appropriate secondary antibodies 1:500 in blocking buffer. For example, when using the vGlut1 guinea pig antibody, apply a secondary antibody that recognizes the guinea pig component (e.g., donkey anti-guinea-pig) conjugated to a specific fluorophore. When using the PSD-95 rabbit antibody, apply a secondary antibody that recognizes the rabbit component (e.g., donkey anti-rabbit) conjugated to a different specific fluorophore.
- Incubate the slices in this solution for 2 – 3 h at RT. Ensure that the slices are protected from the light, as secondary antibodies are light sensitive.
- Wash the slices three times in PBS (10 min each).
- Carefully remove the slices from the 24-well plate using a paintbrush and place them evenly onto pre-labelled glass slides. Add a drop of mounting medium on top of each slice and then gently place a glass coverslip on top of the slices. Avoid the formation of air bubbles. Take care to use enough mounting medium (300-400 µL), as an insufficient amount may lead to dry slices.
- Leave to dry for a minimum of 1 – 2 days at RT and keep the slides protected from light.
- Store the slides at 4 °C in the short-term, but for long-term storage, keep them at -20 °C.
3. Confocal Image Acquisition and Analysis
- Imaging using confocal laser scanning microscopy.
- Identify the region of the hippocampus to be imaged (e.g., the cornu ammonis 1 and 3 (CA1, CA3) or the dentate gyrus (DG)) by means of a 10x or 20x objective.
- Change to a 40x or 63x oil-immersion objective to make sure the slice anatomy is intact by identifying continuous neurites and organized structure. Use a neuronal marker, such as MAP2, as a reference (Figure 1A).
- Subsequently, switch to a 60x oil-immersion objective (NA = ~1.3 – 1.4) and adjust the settings for each channel to obtain optimal signal and contrast. Set the intensity of each laser to avoid the saturation of any pixels using a high-/low-contrast option.
NOTE: These setting will depend on the microscope used, but refer to the Table of Materials for the laser power settings used in Figure 2, Figure 3, and Figure 4. For best results, use a 1024 x 1024-pixel resolution. The settings should remain constant throughout the different conditions of the same experiment. Additional zoom can be applied if required. - Evaluate the depth where the staining is even and acquire image stacks of at least 8 equidistant (250 nm) planes. Then take 3 adjacent representative images stacks from the same area of interest per slice.
NOTE: The depth may vary from one slice to another but should always be approximately 2 – 5 µm from the surface of the slice. - Repeat the acquisition in at least 3 slices per condition and from 6 – 8 animals per treatment group.
- Image analysis.
NOTE: Use an automated image analysis software (See the Table of Materials). Note that some systems require a license, whereas others are free, such as ImageJ. The software used must analyze the images in 3D by taking into account all planes of the stack imaged. Refer to the Table of Materials for the details of the software used in the image analysis (Figure 1).- Use a custom-based intensity threshold protocol to identify all synaptic puncta and neuronal markers, which are manually optimized and selected within the software [see Table of Materials for the specific software used in this protocol].
NOTE: The software identifies a minimum and maximum intensity for each fluorophore and associates a percentage of intensity for each recognized object. Note that the percentage of intensity will vary according to the fluorophore used and the antibody against the synaptic marker. - For synaptic markers, ensure that size filters (>0.1µm3 and <0.8µm3) are selected within the software and applied to the image. Adjust the parameters to ensure that the selected objects are not overlapping. Exclude any object that are too large or too small to be considered synaptic puncta (see Figure 2B).
NOTE: Once optimized, the same threshold values are then applied to all images within a given experiment. - Quantify the mean number, intensity, and volume of individual synaptic puncta (pre- or post-synaptic) using the optimized threshold protocols selected within the software. Normalize the puncta number to the volume of the image field (roughly 16,928 µm3 in the system used here). Normalize the synaptic puncta intensity to the intensity of the reference neuronal marker.
NOTE: Synapses are defined as the co-localization between both pre- and post-synaptic markers. Once the threshold protocols for pre- and post-synaptic puncta have been established, the software should identify the co-localization of synaptic puncta as the overlap of 1 pixel or more in a single plane.
NOTE: For statistical analysis, confirm that all sets of samples follow normality and homogeneity of variance, as determined by Lilliefords and χ2-tests, respectively. Samples showing a normal distribution and homogeneity of variance should be analyzed with parametric tests, as described below. For example, excitatory synaptic puncta analysis should be performed using a one-way ANOVA with blocking and replication. Each individual experiment should be considered as a block; typically, there are three independent experiments, each with 1 – 3 mice from each condition.
- Use a custom-based intensity threshold protocol to identify all synaptic puncta and neuronal markers, which are manually optimized and selected within the software [see Table of Materials for the specific software used in this protocol].

Figure 1: Analysis of synaptic puncta using the image analysis software. (A) Screenshot of the intensity threshold protocol customization. The example given is for PSD-95, in which the selected puncta have a defined size of >0.1 µm3 and <0.8 µm3 and present minimized overlapping objects. Note that the image shown is for one confocal plane to enable the easier visualization of synaptic puncta and accurate parameter selection. (B) Screenshot of the analysis output from the software (following the selection of the optimized protocol). An example of raw synaptic puncta number measurements based on the volume. These values are then exported to a spreadsheet software. Please click here to view a larger version of this figure.

Figure 2: Quality check of the slice and parameter settings for synaptic puncta. (A) Representative confocal images (40x objective) of the region of interest: the CA1 stratum radiatum (sr) and stratum oriens (so) of a hippocampal slice are identified by MAP2 staining. Scale bar = 2 µm. (B) Confocal images (60x objective and an additional 1.3X magnification on the acquisition software) of the CA1 stratum radiatum, with excitatory markers-vGlut1 (green) and PSD-95 (red)-from hippocampal slices. Note the identification of clear pre- and post-synaptic puncta. Puncta larger than 0.8 µm3 are excluded from the quantification (white arrows). Scale bar = 2 µm (C) Confocal images (60x objective and an additional 1.3X magnification on the acquisition software) of the CA1 stratum radiatum, with excitatory markers-vGlut1 (green)-from cryostat-cut hippocampal sections from entire brains fixed in PFA. Note the presence of large holes and irregular, clumped vGlut1 staining. Scale bar = 2 µm. Please click here to view a larger version of this figure.
Synapse loss occurs early in neurodegenerative diseases such as AD and Parkinson's disease2,11,12. However, the molecular mechanisms underlying these deficits remain poorly understood. Deficiencies in Wnt signaling have been associated with AD13,14,15,16,17. Wnts are secreted glycoproteins and play an essential role in synapse formation and the modulation of synaptic transmission18,19,20,21. Recently, we identified Wnts as key regulators of synaptic maintenance in the mature nervous system10,22. To accurately study the impact of Wnt signaling on synapses in the hippocampus of genetically modified mice, we took advantage of a brain slice preparation and confocal microscopy to evaluate changes in synapse number. We identified healthy slices in which the structure was well preserved (Figure 2A). Moreover, the selection of synaptic puncta was carefully defined, with the exclusion of large stained objects probably representing aggregated proteins (Figure 2B). This criterion was applied to all images, with the same strict analysis parameters.
We used a transgenic model system that induces the expression of the secreted Wnt antagonist Dkk1 in adult mice (iDkk1) under tetracycline control (see the Table of Materials)10,22. We demonstrated that the blockade of Wnt signaling in the adult hippocampus triggers excitatory synapse loss specifically in the CA1 stratum radiatum region (Figure 3). Figure 3A illustrates representative confocal images of excitatory pre- and post-synaptic markers (vGlut1 and PSD-95, respectively) acquired from hippocampal slices of iDkk1 mice expressing Dkk1 for two weeks, as well as their respective controls. We analyzed these images using Volocity software and demonstrated a significant reduction in the number of excitatory synapses, quantified by the co-localization of vGlut1- and PSD-95-labeled puncta in the CA1 region of the hippocampus of iDkk1 mice following the induction of Dkk1 for two weeks (Figure 3B, *p <0.05; Kruskal-Wallis test; 6 mice per genotype). In the CA1 stratum radiatum, the excitatory synapse puncta number per 1,000 µm3 was reduced from 500 in control mice to 300 in iDkk1 mice. In contrast, induced Dkk1 expression did not affect the number of inhibitory synapses (identified by the co-localization between the pre- and post-synaptic markers vGAT and gephyrin, respectively), as shown in Figure 4A-B (p >0.05; Kruskal-Wallis test; 3 mice per genotype).
Importantly, we performed a statistical power analysis to estimate the appropriate number of slices and animals required to generate these robust results. Using R, we calculated that approximately 35 slices per condition were needed (3 images x 3 slices x 6 animals = 36) to detect a large effect size (f = 0.4) with 80% confidence (power = 0.8) in a one-way ANOVA with blocking and replication, as described in step 3.2.6.
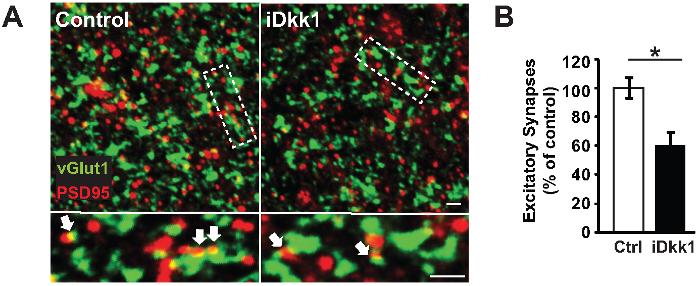
Figure 3: Loss of excitatory synapses in hippocampal slices from iDkk1 mice. (A) Representative confocal images of the CA1 stratum radiatum show excitatory synapses, identified by co-localization between vGlut1 and PSD-95 (white arrows). The lower panel represents a higher magnification of the highlighted rectangle. Scale bars = 2 µm. (B) The percentage of excitatory synapses between control and iDkk1 was quantified using software mentioned in Table of Materials (*p <0.05; Kruskal-Wallis test; 6 mice per genotype). The data are represented ± SEM. This figure has been modified from reference10. Please click here to view a larger version of this figure.
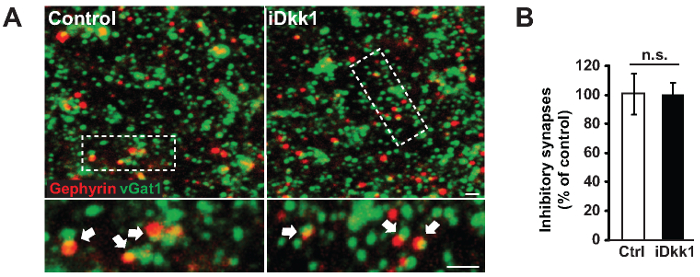
Figure 4:Inhibitory synapses are unchanged in hippocampal slices from iDkk1 mice. (A) Representative confocal images of the CA1 stratum radiatum of inhibitory synapses identified by the co-localization between vGAT and gephyrin (white arrows). The lower panel represents a higher magnification of the highlighted rectangle. Scale bars = 2 µm. (B) Percentage of inhibitory synapses between control and iDkk1 mice, as quantified using Volocity software (p >0.05; Kruskal-Wallis test; 3 mice per genotype). The data are represented ± SEM. This figure has been modified from reference10. Please click here to view a larger version of this figure.
| High Sucrose ACSF Components | Concentration | Notes |
| NaCl | 75 mM | |
| NaHCO3 | 25 mM | |
| KCl | 2.5 mM | |
| NaH2PO4, 2H2O | 1.25 mM | |
| Kynurenic acid | 1.25 mM | |
| Pyruvic acid | 2 mM | |
| EDTA | 0.1 mM | |
| CaCl2 | 1 mM | Add after oxygenation of solution |
| MgCl2 | 4 mM | Add after oxygenation of solution |
| D-Glucose | 25 mM | |
| Sucrose | 100 mM | |
| Submerged chamber ACSF Components | ||
| NaCl | 125 mM | |
| NaHCO3 | 25 mM | |
| KCl | 2.5 mM | |
| NaH2PO4, 2H2O | 1.25 mM | |
| CaCl2 | 1 mM | Add after oxygenation of solution |
| MgCl2 | 1 mM | Add after oxygenation of solution |
| D-Glucose | 25 mM |
Table 1: ACSF composition.
| Blocking/permeabilizing Buffer | Concentration | Notes |
| Donkey serum | 10% | |
| Triton-X | 0.50% | |
| PBS | 1x | |
| 10x PBS | pH to 7.4 | |
| NaCl | 1.37 M | |
| KCl | 27 mM | |
| NaH2PO4 | 100 mM | |
| KH2PO4 | 18 mM | |
| 4% PFA/4% Sucrose | pH to 7.4 | |
| PBS | 1x | Dilute from 10x |
| PFA | 1.33 M | |
| Sucrose | 117 mM |
Table 2: Buffers used for immunofluorescent staining.
