In this protocol, we outlined an updated method to image and analyze initial Ca2+ microdomains in primary mouse T cells based on previous work by our group1,13. This approach was instrumental in unraveling the involvement of CRAC channels such as ORAI1, STIM1, and STIM2, as well as intracellular Ca2+ release channels like RyR1 in early Ca2+ signaling events4.
To do so, we investigated spontaneous Ca2+ microdomain formation by imaging non-stimulated primary mouse Orai1-/-, Stim1-/-, Stim2-/-, and Ryr1-/- and compared them to WT primary mouse T cells. The analysis of Ca2+ microdomain formation encompassed signal onset velocity, Ca2+ amplitude, and number of signals per confocal plane. Notably, except for Stim2-/- T cells, all KO T cells displayed a significant decrease in local Ca2+ signals and a reduced basal free cytosolic Ca2+ concentration compared to WT cells. This led us to conclude that the formation of Ca2+ microdomains is intricately linked to the interaction of ORAI1, STIM1, and RyR14. In addition, we successfully identified and characterized spontaneous Ca2+ microdomains at the plasma membrane. These Ca2+ microdomains were characterized by a Ca2+ amplitude of 290 nm ± 12 nm. Utilizing a color-coded approach for Ca2+ signals allowed for the visualization of Ca2+ microdomains across the cell. Results further highlighted the rapid onset of Ca2+ microdomains, visible within milliseconds, and the capability of this method to detect Ca2+ signals with a longevity of a few milliseconds4. These spontaneous Ca2+ microdomains were later identified as adhesion-dependent Ca2+ microdomains (ADCM), dependent not only on SOCE but also acting via the FAK/PLC-γ/IP3 signaling cascade10 and involvement of P2X49. Furthermore, this technique was fundamental in confirming dual oxidases 1 and 2 (DUOX1/2) as NAADP-producing enzymes21 and HN1L/JPT222 as one of the newly discovered NAADP-binding proteins23.
Figure 2 shows representative examples of Ca2+ microdomains in primary CD4+ T cells upon bead contact from WT as well as P2x4-/- and P2x7-/- mice. Cells were loaded with the Ca2+ dyes Fluo-4 AM and Fura Red AM and imaged at an acquisition rate of 25 ms (40 frames/s). To mimic T cell synapse formation, cells were stimulated with anti-CD3/anti-CD28 coated beads. Initial Ca2+ microdomain formation was analyzed 1 s prior and up to 15 s after bead contact using the DARTS pipeline. Upon bead contact, the WT cell showed rapid formation of Ca2+ microdomains in the first second after stimulation at the bead contact site (Figure 2A). These Ca2+ microdomains further expanded throughout the cell in the following 15 s after bead contact. Opposed to the WT cell, the P2x4-/- and P2x7-/- cells (Figure 2B) showed decreased Ca2+ microdomain formation upon bead stimulation, as well for the P2x4-/- a lower basal level before bead contact. These representative findings are in line with the previously published results by Brock et al.9, indicating Ca2+ microdomain formation in WT T cells directly after bead contact over 15 s and lower signals per frame in P2x4-/- and P2x7-/- cells. Moreover, the amplitude in P2x4-/- cells was significantly reduced, further establishing the role of purinergic signaling in adhesion-dependent Ca2+ microdomains.
Furthermore, this method can also be used to visualize initial Ca2+ microdomains in primary human CD4+ T cells (Figure 3). In line with primary murine T cells, initial Ca2+ microdomains are evoked at the bead contact site. However, the overall Ca2+ response appears to occur on a different timescale compared to murine CD4+ T cells.
The analysis of local Ca2+ signals in a manual manner is not feasible as it is quite laborious and subjective to the individual investigator. Therefore, we previously developed an algorithm in MATLAB Simulink using its image processing and optimization toolboxes for postprocessing13 for analyzing local Ca2+ microdomains.
Recently, we developed a new, open-source, postprocessing pipeline called DARTS for Ca2+ microdomain analysis in high-resolution live cell imaging using the software platform Python12. Here, different deconvolution algorithms can be selected, depending on the user's preference, a cell shape normalization performed to compensate for morphological cell shape changes, and microscope and measurement specific parameters defined (e.g., scale, frame rate, time measured) (Figure 4).
After selecting parameters for Ca2+ microdomain analysis, a second pop-up window is opened for each individual measurement to define the bead contact (Figure 5). To define the bead contact, the user can manually scroll through the tiff file using the slider and select the bead contact frame individually. Bead contact is selected by clicking at the bead contact site (Figure 5, bead and bead contact indicated by yellow ring and arrow) as well as cell selection. This step must be repeated for each cell of interest. Finally, the automated image postprocessing is applied and the results data is summarized and saved in a spreadsheet.
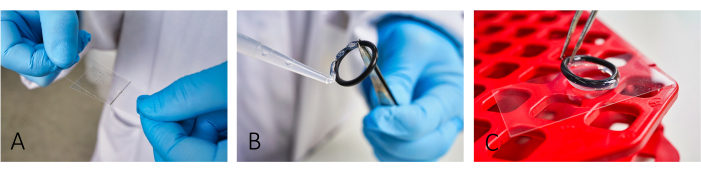
Figure 1: Workflow of slide preparation for imaging. (A) Add and spread both BSA and PLL on the slide using a second glass cover slip. (B,C) To build a chamber, glue the rubber o-rings using silicon grease onto the slide. Ensure that the whole ring is covered with a thin layer of grease to have proper isolation of the chamber. Please click here to view a larger version of this figure.
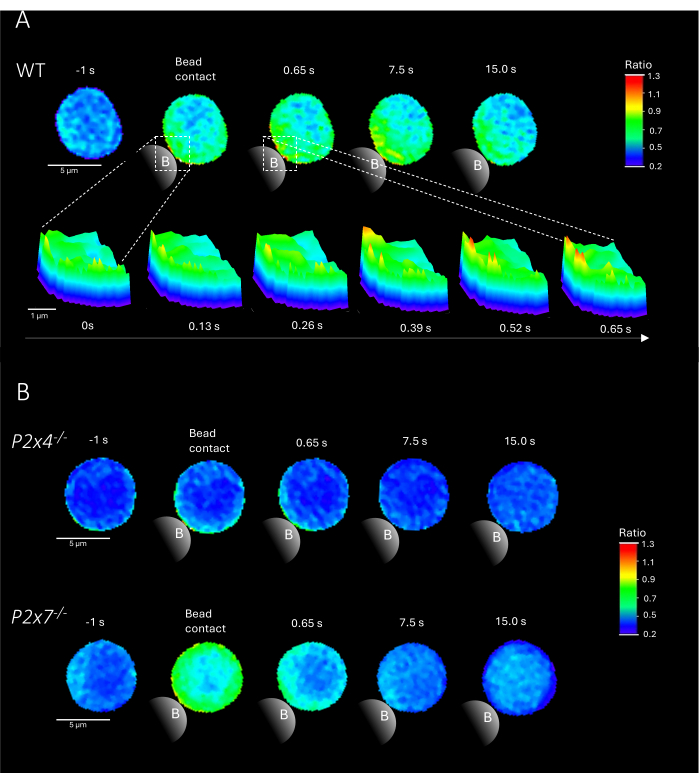
Figure 2: Representative cells of T cell receptor-dependent Ca2+ microdomains in a primary murine wild type (WT) (A), P2x4-/- or P2x7-/- (B) CD4+ T cell. CD4+ T cells were negatively isolated and loaded with Fluo-4 AM and Fura Red, as described above. T cells were analyzed using the DARTS pipeline, resulting in comparable cell images to previously published results9. (A) WT primary T cell 1 s before stimulation with anti-CD3/anti-CD28 coated beads and up to 15 s after stimulation (scale bar 5 µm), as well as 3D surface plot of a zoom-in from 0 s to 0.65 s in the bead contact region (scale bar 1 µm). (B) Upper lane: representative P2x4-/- primary T cell 1 s before and up to 15 s after bead stimulation. Lower lane: representative P2x7-/- primary T cell 1 s before and up to 15 s after bead stimulation. Please click here to view a larger version of this figure.
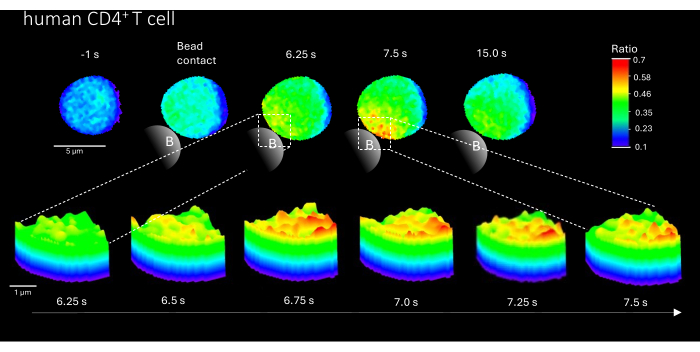
Figure 3: Ca2+ microdomains in a representative primary human T cell after TCR stimulation. Primary human CD4+ T cells were isolated from peripheral blood mononuclear cells (PBMCs) by fluorescence-activated cell sorting (FACS) from buffy coats and loaded with Fluo-4 AM and Fura Red, as described above. The figure shows a primary human T cell 1 s before stimulation with anti-CD3 coated beads and up to 15 s after stimulation (scale bar 5 µm), as well as 3D surface plot of a zoom-in from 6.25 s to 7.5 s in the bead contact region (scale bar 1 µm). Please click here to view a larger version of this figure.
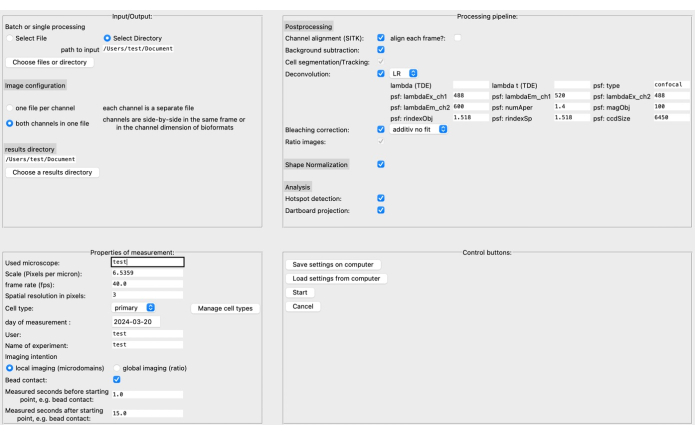
Figure 4: The DARTS graphical user interface (GUI). The GUI is divided into four areas. In the Input/Output area, you need to provide information about the raw data, including the source directory and image configuration (either two channels per file or separate channels), as well as the results directory. In the Properties of Measurement area, the experiment needs to be described with all its relevant information, such as scale (microns per pixel), frame rate, and measurement interval relative to the later determined starting point. Next, a processing pipeline consisting of postprocessing steps, shape normalization, and the actual analysis (microdomain detection and dartboard data accumulation) can be assembled. Finally, the settings can be saved to or loaded from the computer. Once the analysis has been set up, click on Start to proceed. To read more about the setup, visit https://ipmi-icns-uke.github.io/DARTS/General/Usage.html. Please click here to view a larger version of this figure.
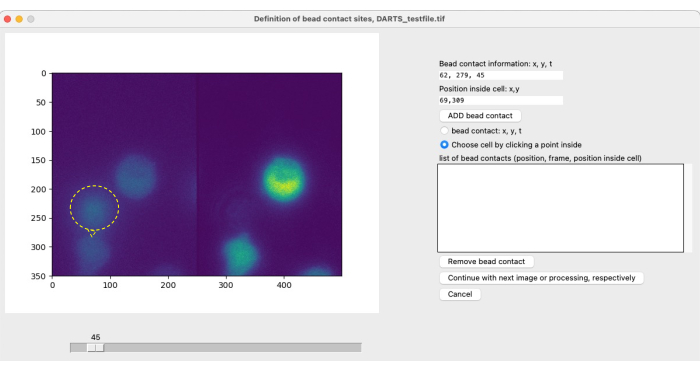
Figure 5: Manual definition of bead contacts. If beads are added to the cells during the experiment, the initial bead contact time with a cell of interest and the contact location have to be manually defined. This is done by scrolling through the frames with the slider and finding a position (x,y) at a time point t. To automatically fill the bead contact information field, the user clicks on the left half of the microscope image at the bead contact position. Next, to associate a cell with the bead contact, the user clicks on a position within the cell that has a bead contact. The information has to be confirmed by selecting ADD bead. Please click here to view a larger version of this figure.
