1. Cloning microRNA Response Elements into the Viral Genome
- Design microRNA response element inserts.
- Identify the desired microRNA and its corresponding target sequence. Several databases are available with mature microRNA sequences. Recommended: http://www.mirbase.org/9,27,28,29,30.
- Clone the response element into plasmid DNA encoding the vector genome or transcript.
NOTE: For two or more copy response elements using a unique restriction site for insertion is easier and more versatile.- Insert response element or unique restriction site using splice-overlap extension (SOE) PCR. This method has been described in detail31.
- If using a restriction site for insertion, purchase commercially synthesized, PAGE-purified sense and antisense ultramers encoding the insert sequences flanked by the overhang sequences of the unique restriction site (Figure 2).
- Combine 0.5 µg sense ultramer, 0.5 µg antisense ultramer, DNA ligation buffer to final concentration of 1x, and bring to 50 µl with H2O. Anneal the ultramers by incubating the reaction at 85 °C for 10 min and then reducing the temperature by 0.5 °C every 30 sec until the reaction reaches 25 °C.
- Combine 0.5 µg of vector DNA encoding the new unique restriction site, enzyme buffer to a 1x final concentration, 1 µl of the appropriate restriction enzyme, and bring to a final volume of 20 µl with H2O. Digest the vector at 37 °C for 2 hr. Purify the linearized DNA by agarose gel purification32.
- Ligate the annealed ultramers into the digested vector overnight at 16 °C using a 3:1 ultramers:vector molar ratio and standard ligation techniques33.
NOTE: When using a single restriction enzyme site for insertion it can be more efficient to dephosphorylate the ends of the digested vector and phosphorylate the ends of the annealed oligonucleotides. - Transform the ligated DNA into E. coli using the heat shock method34.
NOTE: Plasmids encoding viral genomes are generally large, therefore using competent cells optimized to uptake large DNA constructs may increase transformation efficiency. - Purify plasmid DNA from individual colonies using a commercially available purification kit35. Identify an appropriate clone with the microRNA response element in the correct orientation (Figure 2) by sequencing the insert region36.
2. Rescuing microRNA-targeted Picornavirus from Plasmid DNA
- Rescue microRNA-targeted picornavirus in a cell line permissive for virus replication that does not express the cognate microRNA(s). This protocol uses H1-HeLa cells because they do not express miR-142, miR-124, or miR-125, however it is not required to use H1-HeLa cells for rescue.
CAUTION: All guidelines for the safe handling and disposal of infectious agents should be followed accordingly.- Plate H1-HeLa cells in a 6-well plate such that they are ~80% confluent at the time of transfection (24 h post seeding). Plate cells in 2 mL per well of DMEM supplemented with 10% fetal bovine serum.
- Warm the transfection reagent to room temperature. Combine 250 µL serum-free media, 2.5 µg of plasmid DNA encoding microRNA-targeted genome, and 7.5 µL of transfection reagent in a sterile microcentrifuge tube and pipet gently to mix. Incubate the mixture at room temperature for 15-30 min.
- Aspirate the media from the plated cells and add 2 mL of fresh complete media.
- Add the entire transfection mixture from step 2.1.2 to one well of the 6-well plate drop-wise. Add each drop to a different area of the well.
- Incubate the cells at 37 °C until cytopathic effects (CPE) or reporter proteins are detectable (~24-72 h).
- Harvest and passage the rescued virus onto fresh cells.
- Plate cells in a 6-well plate such that they are 80-90% confluent at the time of infection (24 h post seeding).
NOTE: Scale up or down depending on the amount of virus needed for all subsequent experiments. - Harvest each rescue well by scraping the cells into the supernatant using a rubber cell scraper or rubber policeman. Gently drag the scraper across the bottom of the well. Transfer the cells and supernatant into a cryogenic storage tube.
- Freeze/thaw the samples two times and then pellet the cellular debris by centrifuging at 1,200 x g for 5 min at 4 °C.
- Filter the cleared supernatant twice using 0.2 µm syringe filters. The filtered rescue supernatant contains the virus.
NOTE: Filter pore size may vary depending on the size of virus particles and may not be applicable for large viruses. - Aspirate media from the plated cells, wash once with serum-free media and add 1 ml of fresh serum-free media to each well.
- Add 50 µL per well of filtered rescue supernatant and gently rock the plate to evenly distribute virus.
- Incubate the plate at 37 °C for 2 h and then aspirate media from each well to remove unincorporated virus.
- Add 1.5-2 mL of fresh complete media per well and incubate at 37 °C until CPE or reporter proteins are apparent (~24-48 h).
- Repeat steps 2.2.2 to 2.2.4. Filtered rescue supernatant contains virus stock. Store virus stocks in single-use aliquots in cryogenic storage tubes at -80 °C.
- Plate cells in a 6-well plate such that they are 80-90% confluent at the time of infection (24 h post seeding).
3. Rescuing microRNA-targeted Virus from In Vitro Transcribed RNA Transcripts
- Generate RNA transcripts from plasmid DNA encoding the microRNA-targeted viral genome using an upstream promoter.
NOTE: Standard practices for generating and maintaining a nuclease-free environment should be used for all subsequent steps. - Linearize plasmid DNA using an enzyme restriction site downstream of the transcript. Combine 5 µg plasmid DNA, 1x final concentration enzyme buffer, 3 µl restriction enzyme and bring to 50 µl with H2O. Incubate the reaction at 37 °C for 3 hr.
- Add 1/20th volume of 0.5 M EDTA, 1/10th volume 5 M NH4 acetate, and 2 volumes of 100% ethanol to the digested reaction and mix. Incubate at -20 °C for 1 h up to overnight.
- Pellet the precipitated DNA for 10 min at 17,000 x g at 4 °C. Pour off the supernatant, resuspend the pellet in 500 µL of cold 70% ethanol and pellet the DNA by centrifuging at 17,000 x g for 10 min at 4 °C.
- Pour off the supernatant and centrifuge again for 30 sec. Remove the residual supernatant with a pipet and air-dry the pellet. Resuspend the DNA pellet in sterile nuclease-free H2O at a concentration of 0.5-1 µg/µL.
- Thaw the in vitro transcription reagents at room temperature and place the ribonucleotides on ice (enzymes in glycerol do not freeze and should go directly on ice). Keep the reaction buffer at room temperature.
- Assemble the transcription reaction in a PCR tube by combining 2 µL each of ATP, CTP, GTP, and UTP solutions, 2 µL 10x reaction buffer, 1 µg linearized DNA, 2 µl enzyme, and bring to a final volume of 20 µL with nuclease-free H2O.
- Incubate the reaction at 37 °C for 2 hr.
- Purify the RNA transcripts.
NOTE: Before starting, prepare wash solution with ethanol as directed and preheat elution solution to 95 °C for elution step.- Bring the transcription reaction to 100 µL with the elution solution (not-preheated). Add 350 µL of binding solution concentrate and 250 µL of 100% ethanol to reaction and mix.
- Transfer the sample to a filter cartridge inserted into a collection tube and centrifuge for 1 min at 12,000 x g. Discard the flow-through.
- Add 500 µL of wash solution to the filter cartridge and centrifuge for 1 min at 12,000 x g. Discard flow-through. Repeat this wash step one more time. Discard flow-through. Centrifuge for 1 min at 12,0000 x g to remove residual ethanol.
- Transfer the filter cartridge to a clean collection tube and add 50 µL of the preheated elution solution to the filter cartridge. Centrifuge for 1 min at 12,000 x g. Repeat this elution step for a total volume of 100 µl of eluent containing purified RNA transcripts. Discard filter-cartridge.
- Determine the RNA concentration in the eluent by measuring the absorbance of the sample at 260 and 280 nm and using the Beer-Lambert Law.
- Assess the integrity of the RNA using RNA gel electrophoresis37. See Figure 3 for examples of good and bad RNA integrity. RNA should be aliquotted into single-use tubes and stored at -80 °C to maintain integrity.
- Warm transfection reagents to room temperature. Combine 250 µl serum-free media, 2.5 µg of purified RNA transcripts, 5 µL of boost solution, and 5 µL of transfection reagent in a sterile microcentrifuge tube and pipet gently to mix. Incubate the mixture at room temperature for 2-5 min.
- Plate H1-HeLa cells in a 6-well plate such that they are ~80% confluent at the time of transfection (24 h post seeding). Plate cells in 2 mL per well of DMEM supplemented with 10% fetal bovine serum.
- Aspirate the media from the plated cells and add 2 mL of fresh complete media.
- Add the entire transfection mixture from step 3.12 to one well of the 6-well plate drop-wise. Add each drop to a different area of the well.
- Incubate the cells at 37 °C until cytopathic effects (CPE) or reporter proteins are detectable (~24-72 h).
- Continue rescue of microRNA-targeted virus as described in steps 2.2 to 2.2.9.
4. Titrating Virus Stocks by Calculating 50% Tissue-culture Infectious Dose (TCID50)
- Plate H1-HeLa cells in a 96-well plate at 104 cells per well in DMEM supplemented with 10% fetal bovine serum. Incubate cells at 37 °C overnight.
NOTE: Titrate virus in cells permissive for virus replication that do not express cognate microRNAs. - Make 10-fold serial dilutions of virus each in a total volume of 1 mL of serum-free media.
NOTE: Use lower dilutions if more precision is required.
NOTE: Change tips after each dilution to prevent overestimation of titer as virus can stick to tips. - Tip 96-well plate to the side and aspirate media from plated HeLa cells. Add 100 µL of each virus dilution per well to 8-wells of 96-well plate (1 row per dilution). Add 100 µL of serum-free media without virus to row 1 and row 12 on plate for controls.
NOTE: Always aspirate and add media to wells by touching tip to side of well to prevent cell detachment. - Incubate plate at 37 °C for 2 hr. Tip plate to the side and aspirate the media from the wells.
NOTE: Change tips between each dilution row. - Add 100 µl of complete media to each well. Incubate plate at 37 °C for 72 hr.
- Visualize the wells under a microscope and mark each well positive or negative for CPE.
- Calculate each virus titer with the following equation: Log10 (TCID50/ml) = L + D(S-0.5) + log10 (1/V). L is the negative log10 of the most concentrated virus dilution tested in which all wells are positive. D is the log10 of the dilution factor. S is the sum of individual proportions (pi). pi is the calculated proportion of an individual dilution (amount of positive wells/total amount of wells per dilution. V is the volume of inoculum (ml/well).
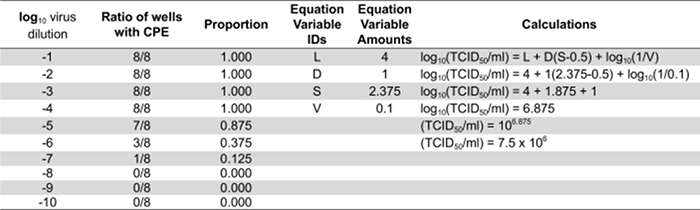
Table 1: Quantifying infectious virus particles by calculating the TCID50. Representative results from cells infected with a series of ten-fold dilutions of a virus stock. "L" equates to 4 because the last dilution where all the wells are positive for CPE is 10-4. "D" is the log10 of 10 since 10-fold dilutions were used and is therefore equal to 1. "S" is the sum of the individual proportions. In this example the individual proportions are 1.0, 0.875, 0.375, 0.125, and 0. The sum of these (S) is 2.375. "V" is the volume of inoculum in ml used to infect the cells initially.
5. Evaluating microRNA-targeting Efficacy: Single-step Growth Kinetics
- Controls for this assay include mock-infected cells, unmodified virus and virus containing a non-targeted or non-functional response element.
- Plate H1-HeLa cells for a time course experiment in 12-well tissue culture plates such that they are 80-90% confluent at the time of infection (24 h post seeding). Plate cells in DMEM supplemented with 10% fetal bovine serum at 1 mL per well.
NOTE: Recommended to plate a separate plate for each time point. This protocol uses 7 different time points for a virus with an 8-12 hr replication cycle. H1-HeLa cells are not required to perform this assay. This assay should be performed in permissive cells that do not express the targeted microRNAs. This assay can also be performed in cells expressing the cognate microRNAs to evaluate growth kinetics under selective pressure. - Aspirate media from wells. Wash wells by adding 0.5 mL of serum-free medium to each well, swirl the plate, and aspirate media from wells. Add 0.5 mL of fresh serum-free media to each well.
- Infect each well at a high multiplicity of infection (MOI; number of infectious particles per cell) to ensure all cells get infected. This protocol uses an MOI of 3.
- Dilute virus stocks in serum-free media to a concentration of MOI = 3 per 100 µL. Add 100 µL of virus dilution each to seven different wells and incubate at 37 °C for 2 h.
- Aspirate the media from all wells and wash two times by adding 0.5 mL complete media per well, rocking gently and then aspirating.
- Add 1 mL of complete media to each well and incubate at 37 °C until desired time point.
- Collect samples for virus titration at 2, 4, 6, 8, 10, 24 and 48 h post-infection (1 well per time point). Transfer 700 µL of the media in the well to a cryogenic storage tube. Scrape the cells into the remaining supernatant by gently moving a rubber scraper across the entire well. Transfer the cell/supernatant mixture to the corresponding cryogenic storage tube containing the 700 µL of supernatant.
NOTE: Make sure not to splash supernatant from one well into another during scraping resulting in contaminated samples. Transferring the 700 µL prior to scraping will minimize splashing. - Place samples at -80 °C until all samples are collected.
- Freeze/thaw the samples three times and remove cellular debris by centrifuging the samples at 1,200 x g for 5 min at 4 °C.
- Titrate the virus as described in section 4 and compare growth kinetics over time.
6. Evaluating microRNA-targeting Specificity: Virus-spreading Assay Using Synthetic microRNA Mimics
NOTE: Use of a synthetic microRNA mimic is performed to show specificity of the microRNA:microRNA-target interaction.
- Include mock transfection, negative control microRNA mimic and experimental microRNA mimic only controls for every assay to evaluate microRNA-mediated toxicity. It is also ideal to include a positive control (i.e. microRNA-targeted gene or genome) as low transfection efficiency of microRNA mimics can result in false negatives.
- Plate H1-HeLa cells in a 96-well tissue culture plate such that they are 80-90% confluent at the time of transfection (24 h post-seeding). Plate cells in DMEM supplemented with 10% fetal bovine serum at 0.1 ml per well.
NOTE: Perform this assay in cells permissive for viral replication that do not express the cognate microRNAs. - Warm the transfection reagents to room temperature. Combine 9 µL serum-free media, 200 nM final concentration per well of microRNA mimic stock, 0.18 µL boost reagent, 0.18 µL transfection reagent and mix. Incubate the mixture at room temperature for 2-5 min.
NOTE: Volumes listed are per well. It is recommended that a master mix be assembled for transfection of all wells to maintain consistency and minimize pipetting error. The optimal concentration of microRNA mimic will vary. Consult the manufacturer's instructions accompanying the microRNA mimic for a reasonable starting concentration. This will generally range from 5-200 nM final concentrations. - Aspirate the media from the wells and add 92 µl of fresh complete medium.
NOTE: If there are a significant number of samples, complete this step prior to assembling the transfection solution and store the plate at 37 °C until ready. - Add the entire transfection mixture in step 6.3 to the cells drop-wise.
- Incubate at 37 °C for 6 h.
- Infect each well at a low MOI to ensure a low percentage of cells get infected to allow analysis of virus spread. This protocol uses an MOI of 0.2.
- Dilute virus stocks in serum-free media to a concentration of MOI = 0.2 per 100 µL. Remove media from the wells and add 100 µL of virus dilution per well.
- Incubate at 37 °C for 2 h.
- Aspirate media from each well and add 100 µL of fresh complete media.
- Incubate at 37 °C for 20-22 h.
- Determine the virus titration in the supernatants.
- Collect the supernatant from each well and replace with 100 µL of fresh complete media.
NOTE: If using suspension cells, resuspend the cell pellet from the following step in 100 µL of fresh complete media and return to sample well for viability assay. - Remove cellular debris from the collected supernatants by centrifuging at 300 x g for 5 min at 4 °C.
- Transfer the cleared supernatant to a fresh tube and titrate infectious virus on permissive cells that do not express the cognate microRNAs as described in section 4.
NOTE: Keep all samples on ice to prevent loss of infectivity.
- Collect the supernatant from each well and replace with 100 µL of fresh complete media.
- Determine the viability of the cells.
- Add 10 µL of MTT (3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide) reagent per well.
- Incubate at 37 °C for 2-4 h until purple precipitate is visible.
- Add 100 µL of detergent reagent.
- Incubate at room temperature in the dark for 2 h.
- Read absorbance of all wells at 570 nm.
NOTE: Normalize all samples to mock transfected cells for comparison of percent cell viability.
Table 1 represents results typical of a titration assay for a picornavirus and describes how to calculate the 50% tissue culture infectious dose. A schematic representation of the overall concept of microRNA-based regulation of viral tropism described in this manuscript is shown in Figure 1. The orientation of microRNA to response element during intracellular interactions, proper design of response element oligonucleotides for annealing and plasmid insertion, and a map of plasmid DNA encoding a microRNA-targeted viral genome for in vitro transcription is depicted in Figure 2. Figure 3 shows RNA transcripts at varying degrees of integrity visualized by agarose gel electrophoresis. Proper handling and the use of DNA templates devoid of impurities will result in properly transcribed RNA transcripts as shown in 3A lane 2. Residual impurities within the linearized DNA templates or trace amounts of nuclease can result in low levels of improperly transcribed RNA and/or degradation products similar to that observed in 3A lane 3. Incomplete linearization of plasmid DNA or DNA containing high amounts of impurities such as residual ethanol will result in improper transcription and degradation products represented in 3A lanes 4 and 5. Figure 3 also shows an image of Mengovirus-mediated cytopathic effects (CPE) following transfection of clean RNA transcripts. Figure 4 displays data representative of a time course experiment evaluating the growth kinetics of unmodified and microRNA-targeted Mengovirus. The results show that microRNA response elements engineered into the Mengovirus genome do not alter the kinetics of virus replication in H1-HeLa cells, which do not express any of the cognate microRNAs. However, in RAW 264.7 macrophages, which express intermediate levels of microRNA-125 and high levels of microRNA-142, viruses encoding the corresponding response elements display inhibited replication kinetics that correlates with the level of microRNA expressed. Data in Figure 5 shows the specificity of microRNA-based regulation of Mengovirus tropism. Overexpression of each individual microRNA in H1-HeLa cells specifically inhibits propagation (lower virus titers) and cytotoxicity (increased cell viability) of Mengovirus encoding the cognate microRNA response element.
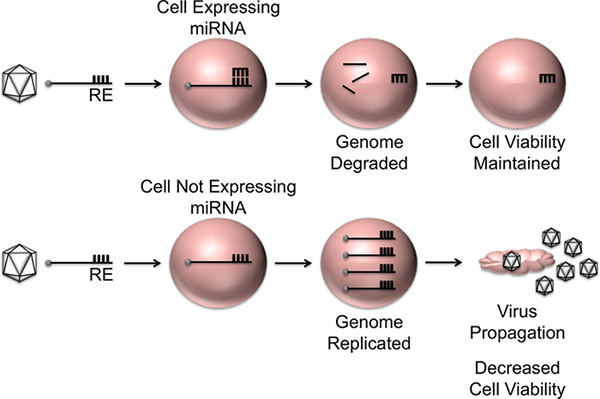
Figure 1: Schematic representation of microRNA-based regulation of viral tropism. MicroRNA response elements (RE) incorporated into a viral genome will be recognized by cognate microRNAs enriched within specific cell types and result in targeted degradation of the viral transcripts/genomes. This will prevent virus replication, spread and toxicity to the surrounding cells. However, viral replication will be maintained in cells that do not express the cognate microRNAs allowing for virus propagation, spread and cell death. Please click here to view a larger version of this figure.
Figure 2: MicroRNA response element design. (A) For successful targeting the seed sequence region of the RE should be perfectly complementary. The RE should be oriented within the viral genome such that it can be recognized by the cognate mature microRNA in mRNA transcripts and/or the viral genome. The majority of REs are placed within the 3' UTR of the transcripts. (B) Depiction of properly designed and annealed oligonucleotides encoding REs containing tandem repeats of the microRNA-target sequences (miRT) flanked by the overhanging nucleotides of the XhoI restriction enzyme site. When designing tandem repeat REs, be sure to include spacer nucleotides (generally 4-6) between each microRNA-target copy. (C) Plasmid DNA encoding a full-length viral genome with a RE incorporated into the 3' UTR, a T7 promoter, and a unique restriction site for linearization for in vitro transcription. Please click here to view a larger version of this figure.
Figure 3: Rescue of virus from genome-encoding RNA transcripts. (A) RNA gel electrophoresis of in vitro transcribed RNAs encoding picornavirus genomes to evaluate transcript integrity. Lane 1: RNA ladder. Lane 2: Properly transcribed RNA with high integrity. Lane 3: RNA transcripts with moderate integrity. Higher band is correct size and lower band is potentially a degradation product or an undesired RNA transcript. Lane 4: Improperly transcribed RNA. Lane 5: Low integrity RNA. 500 ng of in vitro transcribed RNA was loaded per well. Improper transcription or degradation products are likely due to the use of low purity or incompletely linearized DNA. (B) Image of mock transfected H1-HeLa cells and cells transfected with RNA transcripts encoding Mengovirus. Administration of transfection reagents only (Mock) will maintain cell viability following transfection as well as passage onto fresh cells. Transfection of RNA transcripts encoding a Mengovirus genome (vMC24) results in cytopathic effects, which are maintained following filtration of the supernatant and passage onto fresh H1-HeLa cells. Scale bar = 100 µm. Please click here to view a larger version of this figure.
Figure 4: Growth kinetics of unmodified and microRNA-targeted Mengoviruses in the presence or absence of cognate microRNAs. Modified from reference17. (A) H1-HeLa cells do not express miR-124, -125, or -142. All three Mengovirus encoding microRNA REs replicate with similar kinetics as the unmodified virus (vMC24) indicating that insertion of the microRNAs at this location (5' UTR) do not alter virus replication. (B) RAW 264.7 macrophages express intermediate levels of miR-125b and high levels of miR-142-3p, but do not express miR-124. Incorporation of the corresponding microRNA-target sequences into the genome results in virus attenuation consistent with the level of microRNA expression. Data is represented as mean viral titers +/- SD. Please click here to view a larger version of this figure.
Figure 5: Analysis of microRNA-targeting specificity using synthetic microRNA mimics. Modified from reference17. H1-HeLa cells transfected with individual microRNA mimics were infected with unmodified virus (vMC24) or microRNA-targeted virus (miRT-vMC24) at an MOI of 0.2. (A) The cell viability normalized to mock-treated cells was determined at 24 hr post-infection via MTT cell proliferation assay. (B) Virus titer in the supernatant of all samples was also determined at 24 hr post-infection. The data is represented as mean viability or viral titer +/- SD. Two-tailed unpaired Student t tests with Welch's correction (for unequal variances) were used for statistical analysis. A p value <0.01 was considered significant. (**, p <0.01, ***, p <0.001). Please click here to view a larger version of this figure.
