In this study we visualized the bacterial cell division protein FtsZ, expressed in live cells as an FtsZ-GFP fusion, to illustrate the powerful advantages of f3D-SIM over conventional fluorescence microscopy for studying bacterial protein localization and dynamics. FtsZ is a tubulin-like cytoskeletal protein that polymerizes into a dynamic ring structure around the circumference of the cell. The Z ring has an important function in cell division: its assembly marks the future site of division, it recruits all cell division proteins to this site, and Z ring constriction appears to provide the force to allow inward movement of the cell envelope during cytokinesis17,18.
When visualized by conventional wide-field fluorescence microscopy, the Z ring appears as a single transverse band of fluorescence in rod-shaped bacteria, including the most well studied organisms such as B. subtilis (Figure 2A), E. coli, and C. crescentus. Importantly, the fluorescence intensity throughout this band appears to be more or less uniform, and the localization pattern offers very little insight into the structural organization and distribution of FtsZ polymers within the Z ring. When the same cells are examined by using the f3D-SIM method described here (Figure 2B), however, the advantages of this technique are immediately apparent. First, rotation of the 3D-SIM image around the z-axis enables visualization of the Z ring as a genuine ring structure in three dimensional space (Figures 2C and 3A). Together with the increase in resolution, this reveals that the distribution of FtsZ in the Z ring is in fact highly heterogeneous (Figure 3). The improvement in resolution also allows us to see Z rings that have begun the process of constriction to form a division septum (indicated by the invagination of cell membrane in Figure 2C and 2D).
Additional examples of B. subtilis Z rings visualized by this technique are shown in Figure 3Bi – 3Biv and illustrate how the fluorescence intensity around the Z ring is never the same. Moreover, regions of very low fluorescence intensity are often observed. We refer to these regions as gaps (white arrowheads). We observe these gaps in 15% of all Z rings examined (n = 84) and predominantly in Z rings having a diameter of 800 – 900 nm (93%). To quantify these observations, 3D intensity plots were created of the Z ring and are shown in Figure 3Ci and 3Cii using the Z rings shown in Figures 3Biii and 3Biv. The intensity plots show that typically the amount of fluorescence in the Z ring can vary up to 3 – 4 fold in different regions of the B. subtilis Z ring. Furthermore the 3D intensity plots show that the gaps of fluorescence inside the Z ring almost read baseline levels of background fluorescence (black arrow in Figure 3Ci). These above examples depict good reconstructions of the Z ring and are dependent upon sufficient light being emitted from the sample without significant photobleaching. This is accomplished through the brightness of the GFP fluorophore fused to FtsZ and its abundance in the cell under these conditions (high signal to noise ratio). In other cells where the signal to noise ratio is low the reconstruction of the image is poor. To simulate this occurrence with FtsZ-GFP, the amount of emission signal collected from the B. subtilis cells was reduced by lowering the excitation energy. As shown in Figure 3D a 100 fold reduction of excitation energy results in an image of the Z-ring that has significant artifacts. This occurs as a consequence of insufficient local contrast between the FtsZ-GFP signal and the background leading to poor reconstruction of the image.
Interestingly, B. subtilis Z rings showed more pronounced gaps and heterogeneity in the top and bottom regions of the ring but not in the sides of the ring (Figure 3B). This occurs because of the difference in resolution achieved by this variation of 3D-SIM in the lateral plane compared to the axial plane. This results in the top and bottom regions of the Z ring (lateral plane) being imaged with optimal resolution. The gaps in the Z-ring are too small to be detected in the sides of the Z ring as visualized in the axial plane, which has about half the resolution of the lateral plane. Bacteria that have a coccoid morphology, such as S. aureus, have Z rings which are positioned in all planes. So imaging the Z ring when it lies in the lateral plane (or close to it) provides the ability to image all regions of the ring with optimal resolution. Taking advantage of this, we examined the structure of the Z ring in live S. aureus cells, utilizing a strain that contains a plasmid-bearing an ftsZ-gfp fusion. Production of this fusion is under the control of a Pspac inducible promoter (see step 1.2.1). When imaged with in the axial plane, S. aureus Z rings appeared identical to B. subtilis Z rings which are present in the same plane (Figure 4Ai). However, imaging Z rings in the lateral plane confirmed that the entire Z ring appeared both bead-like and heterogeneous. About 26% of these rings also showed at least one visible “gap” of fluorescence (Figure 4Aii, 4B). Similar to B. subtilis FtsZ, fluorescence intensity plots show that the intensity of fluorescence in the Z ring varies as much as 4 – 6 fold in different areas of the S. aureus Z ring (Figure 4C).
The length and width of the beads can be measured (refer to schematic in Figure 4D). This showed that 80% of the Z rings (n = 43) that were examined had a bead length of 200 nm, reaching a maximum length of 400 nm. The width of beads, on the other hand, measured up to 200 nm. There was on average 12 ± 2 (SEM) FtsZ beads per Z ring. Similar localization data was observed when native FtsZ was visualized with these methods using immunofluorescence microscopy (IFM) showing that this heterogeneous and bead-like localization pattern was genuine and not an artifact caused by expression of ftsZ-gfp in addition to native FtsZ (data not shown). This advancement of 3D-SIM is able to show that the Z ring appears as a heterogeneous structure and is possibly discontinuous in nature.
To address the question of how this heterogeneous Z-ring structure undergoes constriction and facilitates cell division, a time-lapse analysis of Z ring dynamics can be performed. One of the main challenges with fluorescence microscopy time-lapse studies is in the minimization of sample photobleaching and phototoxicity during imaging. 3D-SIM requires a large number of raw images to be obtained which means that photobleaching of a sample over time will result in a poor signal-to-noise ratio leading to insufficient signal to allow appropriate image reconstructions. The application of the new technology minimizes the amount of excitation energy used for each exposure and hence a decrease in the energy entering the live cells. It does this by a combination of light beam interferometry to create the structured pattern upon the sample and the use of scientific CMOS cameras which increase sensitivity and quantum efficiency. This combination results in z-stack acquisition of 1 µm per sec (512 × 512 pixels x 8 z-slices) compared to 1 µm per 5-8 sec (512 x 512 pixels x 8 z-slices) obtained with our previous iteration of 3D-SIM (described in Riglar et al.26). Decreasing the image acquisition time and exposure settings can also help minimize phototoxicity of live cells which is particularly relevant in time-lapse studies. The new technology presented here also addresses another problem in live-cell imaging that we refer to as ‘motion blur’ which in this context refers to the localization of proteins changing during image acquisition. By decreasing the image acquisition time the amount of ‘motion blur’ is minimized thus creating a more accurate image reconstruction.
Previous studies that have addressed how FtsZ is organized within the Z ring have not been able to show how Z ring structure changes over time. To investigate this aspect we performed time-lapse using f3D-SIM. This enabled acquisition of a 3D-SIM image of the Z ring in B. subtilis every 5-10 sec over a period of 50 sec, which was previously not possible on this time scale. Shown in Figure 5A is an example of the rapid changes in FtsZ distribution within the Z ring over time (diameter of 890 nm). Other Z rings that were visibly in the process of constriction were also shown to have similar dynamics that affected the distribution of FtsZ around the Z ring (not shown). 3D intensity plots can also be generated for each of the time points to quantify and appreciate the continual fluorescence intensity changes and thus relative abundance of FtsZ in different regions of the Z ring on a time scale of seconds (Figure 5B). Regions of interest can be identified from these intensity plots to track the fluctuations of fluorescence intensity (Figure 5C). Tracking these changes in the Z ring structure under these conditions would be virtually impossible without the advanced f3D-SIM technology. In addition, this work revealed that in S. aureus the dynamic movement of FtsZ is similar to that observed in B. subtilis. S. aureus RN4220 cells producing FtsZ-GFP were imaged for 50 sec at 10 sec time intervals. Changes to the heterogeneity of the Z ring in S. aureus with f3D-SIM were also similar to B. subtilis (see arrowheads and arrows in Figure 6). These time lapse studies support a model of Z ring constriction (the iterative pinching model) that was predicted through examination of fixed C. crescentus cells, but could not be demonstrated due to the need to examine Z-ring dynamics in live cells27.
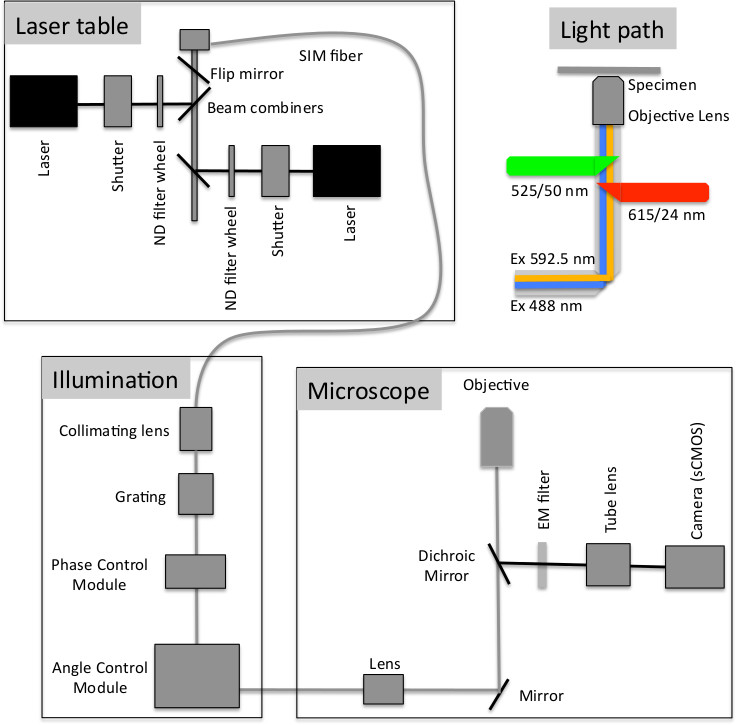
Figure 1. Schematic of the optical configuration of the f3D-SIM used in this study. Laser light from the laser table is directed through a SIM fiber to a collimating lens and fixed grating, then into the phase control module that creates both the wavelength specific optimal spacing of the ± 1st order beams from the zero order central beam and the phase steps for the SIM collection. The beams then enter the angle control module where the beams are reflected to the 3 different clusters to create the 3 angles of illumination. The beams then enter the microscope and light path as shown (Modified with permission from Paul Goodwin, API).
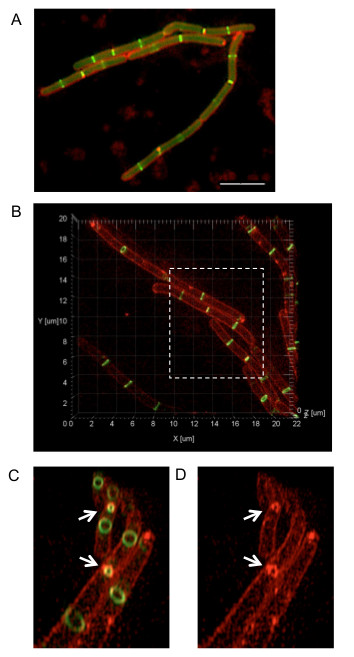
Figure 2. Comparison of conventional wide-field fluorescence and 3D-SIM imaging of B. subtilis cells expressing ftsZ-gfp. (A) Conventional wide-field fluorescence imaging of B. subtilis cells expressing ftsZ-gfp (SU570; green) as a single copy were also stained with the membrane dye FM4-64 (red). Scale bar = 5 µm. The image was acquired using an upright fluorescence microscope. (B) 3D-SIM imaging of the same B. subtilis strain expressing ftsZ-gfp, stained with FM4-64. Regions of interest from the image can be selected (dashed box) to zoom in. The image can also be rotated around the z-axis to view 3D FtsZ rings in the axial plane. (C, D) The removal of out-of-focus light and improved image resolution provided by 3D-SIM allows clear visualization of Z rings (including those that are constricting) and the inner cell membrane during division (indicated by white arrows). Note that the protocol text describes the visualization of a single fluorophore. However, the procedure can be adapted for acquiring up to four different wavelengths simultaneously. This figure has been reprinted from Strauss et al16.
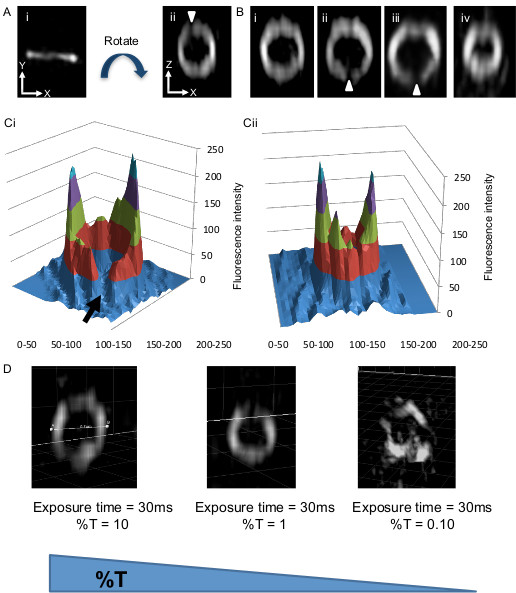
Figure 3. Representative images of the Z ring in live rod-shaped bacterial cells (B. subtilis) expressing FtsZ-GFP using 3D-SIM. (Ai) The Z ring is observed as a band of fluorescence perpendicular to the long axis of the cell which is in the x-y plane. (Aii) The image has been rotated around the z-axis using Imaris 3D visualization software (refer to protocol step 6.1) so that one is looking through the ring structure to clearly see how FtsZ is distributed within the Z ring. This image, now in the z-x plane shows that the ring has a heterogeneous distribution of FtsZ with regions of no or very little fluorescence (white arrowheads). The image must be rotated to observe these ‘gap’ regions of fluorescence. Bi-Biv show further examples of rotated Z rings that now lie in the z-x plane. Z rings in (Bi–iii) have a diameter of ~0.9 µm. (Biv) shows a Z ring with a diameter of only 0.65 µm undergoing constriction. (Ci) This typical 3D intensity profile illustrates the fluorescence intensity (i.e. concentration) differences in FtsZ-GFP around the Z ring having a diameter of 0.85 µm (refer to protocol steps 6.1-6.3). The level of fluorescence in these gaps is low and approaches background fluorescence levels (black arrow). (Cii) A similar 3D intensity profile of a Z ring in the process of constriction. FtsZ-GFP concentration is also non-uniform ; diameter, 0.65 µm. Panels A, B, CI and Cii have been reprinted from Strauss et al16.
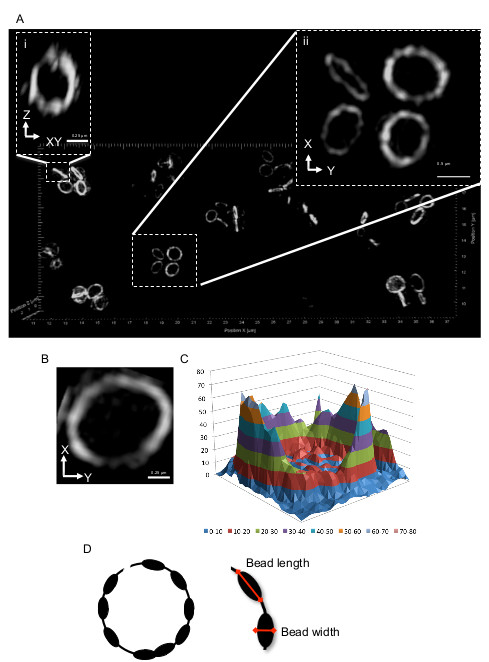
Figure 4. Representative 3D-SIM images of S. aureus cells producing FtsZ-GFP. Since these cells are round (cocci), the Z ring will have various orientations. Cells that show a band of fluorescence in the normal viewing orientation (that is, in the x-y plane) as shown in (Ai), look very similar to those in rod-shaped cells when rotated around the Z axis as for Figure 1Ai. In (Aii) the Z ring is already in the x-y plane; lateral orientation and resolution is maximal over the entire ring which now gives a bead-like structure (B). (C) A 3D intensity plot of the relative abundance of FtsZ-GFP in the Z ring. (D) Schematic representation of the S. aureus Z ring. Red arrows indicate bead length and width dimensions (refer to protocol step 6.1). This figure has been modified from Strauss et al16.
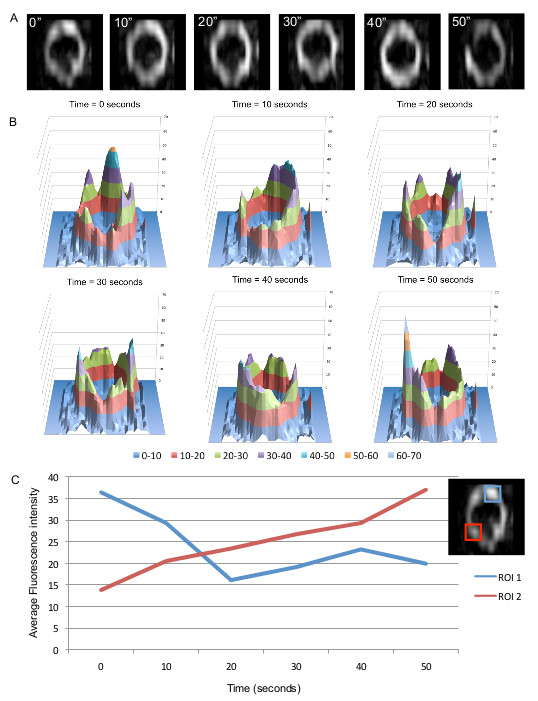
Figure 5. f3D-SIM allows fast analysis of FtsZ localization over time in 3D-SIM. (A) Changes in FtsZ-GFP distribution within the Z ring in rod-shaped B. subtilis cells can be visualized to indicate the dynamics of the ring. Time (sec) is indicated on the upper left corner of each image. (B) 3D fluorescence intensity plots show the non-uniform and dynamic distribution of FtsZ in the Z ring. (C) FtsZ dynamics (FtsZ-GFP fluorescence changes in the ring) can also be examined in more detail by quantifying fluorescence intensity over time in two regions of interest. This figure has been reprinted from Strauss et al16.
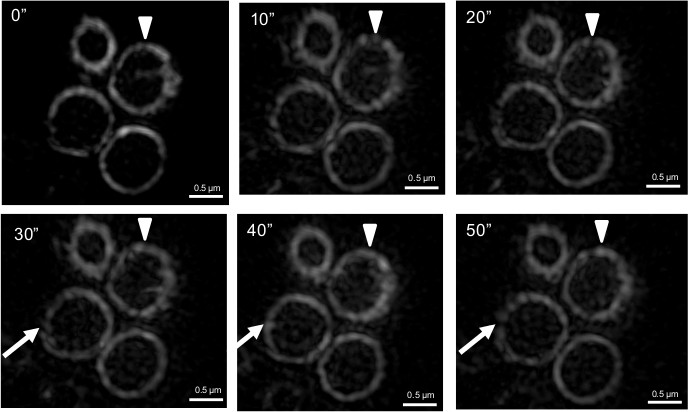
Figure 6. f3D-SIM time-lapse images show changes in FtsZ localization within the ring over time in S. aureus. A white arrowhead indicates a gap when it is initially detected in the Z ring. The appearance and disappearance of gaps at the same position (as marked by the white arrowhead) over time illustrates how FtsZ is redistributed around the Z-ring. White arrows indicate the formation of gaps in the Z ring at later time points. Time (sec) is shown on the upper left-hand side of the images. This figure has been modified from Strauss et al16.
