1. Incorporate Unique DNA Barcodes onto a Plasmid Containing the Necessary Components for Allelic Exchange
NOTE: A new plasmid, named pSKAP, with a high copy number and increased transformation efficiency compared to the existing pKD13 allelic exchange plasmid was created. This is described in steps 1.1-1.12 (Figure 1). The finalized plasmids containing unique DNA barcodes and components for allelic exchange are available through a plasmid repository (Table of Materials).
- Using a commercial plasmid miniprep kit, purify pKD1314 and pPCR Script Cam SK+ from overnight bacterial cultures grown in Luria-Bertani (LB) broth supplemented with 50 μg/mL kanamycin or 25 μg/mL chloramphenicol (for pKD13 and pPCR Script Cam SK+, respectively) (Table 1).
- Perform restriction digestions on both plasmids using commercial restriction enzymes HindIII and BamHI according to the manufacturer’s specifications.
- Remove restriction enzymes and excised DNA from the pPCR Script Cam SK+ reaction and purify the 3,370-base pair (bp) plasmid backbone using a commercially available DNA cleanup kit according to the manufacturer’s specifications.
- Separate the fragments from the pKD13 restriction digestion on a 1% agarose gel using an electrophoresis chamber.
- Visualize bands using a blue light transilluminator and excise the 1,333 bp fragment from the gel (Figure 1C).
NOTE: This fragment contains the FRT-flanked kanamycin resistance gene required for chromosomal allelic replacement. - Purify the excised DNA from step 1.5 using a commercial gel extraction kit.
- To create pSKAP, ligate the purified fragment from pKD13 (from step 1.6) into pPCR Script Cam SK+ (from step 1.3) using a commercial T4 DNA ligase according to the manufacturer’s specifications.
- Transform chemically competent DH5α cells with the ligated pSKAP plasmid following the manufacturer’s protocol (Table 1).
- Spread transformants onto LB agar plates supplemented with 50 μg/mL kanamycin and incubate at 37 °C overnight.
- Pick a colony from the plate and streak it on a new LB agar plate supplemented with 50 μg/mL kanamycin and incubate at 37 °C overnight. Pick a colony from this plate and use to inoculate LB broth supplemented with 50 μg/mL kanamycin. Incubate the culture overnight at 37 °C with constant agitation.
- Use a commercial plasmid miniprep kit to purify pSKAP from the overnight bacterial culture.
- Perform a diagnostic restriction digestion of the plasmid from step 1.11 using Hind III and Bam HI according to manufacturer’s specifications. Visualize fragments on a 1% agarose gel as in steps 1.4-1.5.
NOTE: The pSKAP total size should be 4,703 bp. Fragments after step 1.12 should be 3,370 and 1,333 bp. - Design PCR primers for insertional site-directed mutagenesis (SDM) (Table 2 and S1) in such a way as to insert a unique 25-basepair DNA sequence at position 725 of pSKAP (Figure 2).
NOTE: The barcode DNA is inserted into the plasmid just outside of the FRT-flanked kanamycin resistance gene, so the barcode is not lost during subsequent removal of the kanamycin resistance cassette. If generating new barcode sequences, use online tools to ensure that the fluorescently-labeled target-specific PCR probes (hereafter referred to simply as “probes”) will efficiently bind to the new sequence. Insertion sequences designed to-date, along with the necessary primers to create them, are provided in Tables 2 and S1. - Prepare SDM reactions using a commercial high-fidelity DNA polymerase, the desired primer pairs (Table 2), and pSKAP template. Set the thermocycler to perform the following: 1) 98 °C for 30 s, 2) 98 °C for 10 s, 56 °C for 15 s, 72 °C for 2 min, 3) repeat steps 2, 24 times, 4) 72 °C for 5 min, 5) hold at 4 °C.
- After completion of PCR, deplete pSKAP template by adding the restriction enzyme Dpn I to the reaction. Incubate at 37 °C for 20 min.
NOTE: Products from PCR should be visualized on an agarose gel to verify the size and purity of the product. A control Dpn I digestion consisting of the unmodified pSKAP template can be performed and used in subsequent steps to ensure template DNA is completely digested. - Use 5 μL of the product from step 1.15 to transform 100 μL of commercial chemically competent DH5α cells according to the manufacturer’s recommendations.
- Spread transformants onto LB agar plates supplemented with 25 μg/mL chloramphenicol and incubate at 37 °C overnight.
- Select a colony (or colonies) from overnight plates and streak onto individual LB agar plates supplemented with 25 μg/mL chloramphenicol and incubate at 37 °C overnight. Select a colony from overnight plates and use to inoculate 5 mL of LB broth supplemented with 25 μg/mL chloramphenicol. Incubate culture(s) overnight at 37 °C with constant agitation.
- Use a commercial plasmid miniprep kit to purify plasmids from the overnight culture(s).
- Sanger sequence purified plasmids using the M13 Forward sequencing primer (Table 2). Compare mutated region to the original plasmid and assess for SDM insertional accuracy.
- After confirming the barcode insertion and accuracy, assign each barcode and plasmid a name.
NOTE: Barcodes generated to-date have been assigned a two-letter designation: AA, AB, AC, …, BA, BB, BC, etc. Barcoded plasmids are denoted as pSKAP_AA, pSKAP_AB, pSKAP_AC, …, pSKAP_BA, pSKAP_BB, pSKAP_BC, etc. - Repeat steps 1.14-1.21 to generate the desired number of DNA barcodes.
2. Introduce DNA Barcode onto the Chromosome of S. Typhimurium
NOTE: Insertion of DNA barcodes onto the S. Typhimurium chromosome is achieved by using an allelic exchange method described by Datsenko and Wanner14 that has been modified for use in S. Typhimurium.
- Determine the locus on the S. Typhimurium genome at which to insert the DNA barcode (Figure 3).
NOTE: Select a large, intergenic region of the chromosome. Avoid regions that produce non-coding RNA. This study utilized a locus downstream of put P between residues 1,213,840 and 1,213,861 (determined from genome assembly GCA_000022165.1). This region has previously been genetically manipulated for in trans complementation of genes15. Alternatively, a DNA barcode could be introduced while simultaneously disrupting a gene of interest. Doing so would require minimal alterations to this protocol and streamline mutant creation. - Design PCR primers to amplify the unique barcode and FRT-flanked kanamycin resistance gene from the desired pSKAP barcode-containing plasmid from step 1.21 (Table 2). Add 40-nucleotide extensions that are homologous to the region selected in step 2.1 to the 5’ end of each primer (Table 2).
- Perform the amplification using a commercial high-fidelity polymerase, the primers from step 2.2, and the desired pSKAP barcode-containing plasmid as template. Set the thermocycler to perform the following: 1) 98 °C for 30 s, 2) 98 °C for 10 s, 3) 56 °C for 15 s, 4) 72 °C for 60 s, repeat steps 2-4 29 times, 5) 72 °C for 5 min, 6) hold at 4 °C.
- After completion of PCR, deplete template DNA using DpnI as described in step 1.15. Purify and concentrate the DNA using a commercial DNA cleanup kit according to the manufacturer’s specifications.
- Refer to the previously published protocol for generating mutants in S. Typhimurium strain 14028s from PCR products14,16,17,18.
NOTE: It is not essential that the kanamycin resistance gene is excised from the chromosome. However, excision of the gene is minimally disruptive to the bacterial chromosome as it results in a 129 bp scar that would leave potential downstream genes in-frame. It is recommended that the strain containing the kanamycin resistance gene is retained as it can be used to move barcodes between strains via P22-mediated transduction. - Repeat steps 2.3 – 2.5 to create the desired strains with the appropriate barcodes.
NOTE: Barcodes can be introduced into wild-type S. Typhimurium that can then be subjected to further genetic manipulation, or barcodes can be introduced into strains that have been previously genetically altered.
3. Bacterial Growth Conditions and In Vitro Competition Assays
- From bacteria stock, streak desired S. Typhimurium strains that each harbor a unique DNA barcode onto LB agar plates. Incubate plates overnight at 37 °C.
- Select a single colony from each strain and inoculate 5 mL of LB broth. Incubate for 20 h at 37 °C with constant agitation.
NOTE: Using the overnight culture, proceed to step 3.5 to collect pure genomic DNA (gDNA) from each barcoded strain. This is necessary for subsequent validation and control experiments in section 6. - For each competition assay, transfer an equal volume of each overnight culture into an appropriately sized sterile tube. Thoroughly mix strains together by vortexing vigorously for at least 5 s.
NOTE: The volume of each overnight culture to transfer should be sufficient for each condition and replicate, as well as for isolating gDNA to quantify input. While it is not entirely necessary to measure optical densities of cultures because the absolute number of input microorganisms will be quantified using digital PCR, the number of input bacteria for each strain should be approximately equal to avoid bottlenecks or unequal competition early in the experiment. Representative competition assays in this protocol compared growth rates of 8 strains simultaneously. Additional or fewer strains may be necessary for individual experimental designs. - Transfer 100 μL of the mixed inoculum into 4.9 mL sterile LB broth. Incubate at 37 °C for the desired time or to a desired optical density.
- Harvest 500 μL of the inoculum by centrifugation at >12,000 x g for 1 min. Remove and discard the supernatant. Proceed immediately to section 4 with the cells.
- At desired time points, remove 500 μL aliquots of culture and harvest cells by centrifugation at >12,000 x g for 1 min. Remove and discard supernatant.
NOTE: If collecting aliquots at multiple timepoints, freeze pellets at -20 °C or immediately proceed to step 4.1 after each collection.
4. Collecting and Quantifying gDNA from S. Typhimurium (from Steps 3.5 and 3.6)
- Harvest gDNA from cells using a commercial gDNA purification kit. If available, perform the optional RNA depletion step.
NOTE: RNA depletion is not necessary; however, the presence of RNA will artificially increase the DNA concentration, leading to aberrant calculations in subsequent steps. If using a commercial gDNA purification kit, follow the manufacturer’s recommendations to ensure that the column is not overloaded with DNA. There is no minimum DNA concentration required if the sample is quantifiable in subsequent steps. - Use a spectrophotometer to quantify DNA in each sample.
NOTE: DNA can be quantified using any reliable method. - Calculate gDNA copy number based on the bacterial genome size using the following equation where: X is the amount of DNA in ng and N is the length of a double-stranded DNA molecule (the genome size).
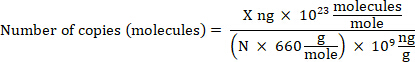
NOTE: 660 g/mole is used as the average mass of 1 DNA bp. Small variations may exist depending on the organism’s nucleotide composition. Numerous calculators are available online to perform the calculation.
5. Design Primers and Probes for Quantitative Detection of DNA Barcodes via dDigital PCR
- Design primers to amplify the barcoded region of the S. Typhimurium chromosome downstream of putP (Figure 3C and Table 2).
NOTE: Primer and probe designs can be facilitated by numerous online programs (Table of Materials). If barcodes are all inserted at the same loci, a single set of amplification primers is universal for all barcodes. - Design 6-carboxyfluorescein (FAM)-based and/or hexachlorofluorescein (HEX)-based probes specific to each barcode (Table 2 and S1).
NOTE: The droplet reader used in this experiment is capable of detecting both FAM- and HEX-based probes simultaneously in a multiplex reaction. Design ½ of the probes to utilize FAM and ½ to utilize HEX. This is not a necessary step but will reduce the reagent use and experimental costs if implemented. - Make 20x primer-probe master mixes containing 1) 20 mM of each forward and reverse amplification primer, 2) 10 mM of a single FAM probe, and 3) 10 mM of a single HEX probe (if multiplexing).
6. Validate the Sensitivity and Specificity of Each Pprimer-probe Set for Each Genomic Barcode Using Digital PCR
NOTE: This protocol uses validating eight unique barcodes with eight unique probes as an example. The number of barcodes utilized can be increased or decreased to accommodate various experimental designs.
- Create a pool of gDNA that contains every barcode except for one. Use this pool as the diluent to perform a dilution series with gDNA containing the single remaining barcode (sample dilution scheme is provided in Figure 4).
NOTE: Using pooled gDNA as a diluent ensures a consistent background while ascertaining sensitivity. Using the copy numbers determined in step 4.3, dilute gDNA to a copy number within the recommended digital PCR range (1-100,000 copies per 20 μL reaction). Keep in mind that the range is set for each unique target (barcode), not the total gDNA. - Prepare reactions for digital PCR in duplicate according to the manufacturer’s recommendations for using a digital PCR supermix designed for probe-based chemistry. Use the mixtures from step 6.1 as template DNA. Use the 20x primer-probe master mix from step 5.3 that contains the probe for the diluted barcode in step 6.1.
- Prepare replicate control reactions for digital PCR according to the manufacturer’s recommendations for using a digital PCR supermix designed for probe-based chemistry. Control reactions for each probe mix must consist of 1) no template controls (NTCs), 2) negative controls, and 3) positive controls.
NOTE: The minimum number of replicate control reactions is two. This example protocol uses four NTCs, six negative controls, and six positive controls for each barcode. Negative controls should contain gDNA with each barcode except for the barcode corresponding to the probe being tested. This will validate the specificity of each probe. - Repeat steps 6.1-6.3 to create digital PCR reactions for each barcoded gDNA sample. A sample plate arrangement is presented in Figure 5.
NOTE: This and subsequent steps are described based on a specific digital PCR platform that utilizes droplets and flow-based technology. Alternative digital PCR platforms that utilize chip-based technology can easily be substituted with slight modifications to this protocol. Step 6.4 may require more than one 96-well plate to validate all primer sets. In contrast to qPCR, separate plates analyzed by digital PCR can be readily compared without the need for standardized reference wells between plates. - Generate droplets for each reaction condition using a droplet generator according to the manufacturer’s instructions.
- Transfer newly created droplets into the appropriate 96-well plate. Use 200 μL pipette tips on a 5-50 μL multichannel pipette.
NOTE: When pipetting droplets, pipette slowly and smoothly! The digital PCR equipment manufacturer recommends using only pipettes and pipette tips from a particular manufacturer (e.g., Ranin). These pipette tips have a smooth opening with no microscopic plastic fragments that can destroy droplets or damage the microfluidics of the droplet reader. Numerous brands of tips were examined and observed to have a spectrum of manufacturing quality. Equivalent results have been achieved using pipette tip alternatives; however, caution should be used when deviating from the manufacturer’s recommendations. - After all the droplets have been generated and transferred, seal the plate with a foil plate sealer.
- Use the manufacturer-recommended thermocycler to perform the following cycling conditions: 1) 94 °C for 10 min; 2) 94 °C for 1 min, ramp rate set at 1 °C/s; 3) 55 °C for 2 min, ramp rate set at 1 °C/s; 4) repeat steps 2 and 3 49 times; 5) 98 °C for 10 min; 6) hold at 4 °C up to 24 h.
NOTE: Thermal transfer in a droplet reaction is not the same as standard PCR. Reaction conditions may require modification. - While thermocycling is being performed, program the data analysis software with the plate setup information such as sample name, experiment type (absolute quantification), supermix used, target 1 name (FAM barcode name), target 1 type (NTC, positive control, negative control, or unknown), target 2 name (HEX barcode name), target 2 type (blank, positive control, negative control, or unknown). The final plate setup information is shown in Figure 5.
- After thermocycling is complete, transfer the completed reactions to the droplet reader and start the reading process according to the manufacturer’s instructions.
7. Quantify the Number of Bacteria in a Competitive Index Experiment
- Dilute gDNA isolated and quantified from section 4 to an appropriate concentration as described above.
- Prepare reactions for digital PCR according to the manufacturer’s recommendations for using the appropriate supermix. Use DNA from step 7.1 as the template. Use one 20x primer-probe master mix that contains the probe (or probes if detecting both FAM and HEX) for possible barcodes present in the experiment.
- Prepare additional digital PCR reactions as in step 7.2 using different 20x primer-probe master mixes until all barcodes utilized in the experimental design can be detected.
- Include controls for each condition as described in 6.3. This includes 1) no template controls (NTCs), 2) negative controls, and 3) positive controls.
- Continue with the protocol as described in steps 6.5-6.10.
8. Analyze Digital PCR Data and Calculate Absolute Copy Numbers
- When all wells have been read and the run is complete, open the .qlp data file using the data analysis software.
NOTE: The file types and data analysis procedures described here are specific to one digital PCR manufacturer. If using alternative digital PCR platforms, file types and data analysis procedures will be specific to the platform used and should be performed according to the manufacturer’s recommended specifications. - Select all wells that utilize the same primer-probe master mix.
- On the Droplets tab, examine the number of droplets analyzed in each well (both positive and negative droplets). Exclude from analysis any well that has fewer than 10,000 total droplets.
- Move to the 1D Amplitude tab and examine the amplitudes of positive and negative droplets. Ensure they comprise two distinct populations.
- Within the software, use the thresholding feature to make a cutoff between positive and negative droplets for each probe that was utilized (Figure 6).
NOTE: All wells that use the same primer-probe master mix from step 5.3 should have the same thresholds. - Once appropriate thresholds have been applied to all wells, the software will calculate the number of DNA copies in each reaction. Export data to a spreadsheet to facilitate further analysis.
NOTE: The data analysis software uses the number of positive and negative droplets that are fit to a Poisson distribution to determine the copy number. - Using the values from step 8.6, calculate the initial copy number of each unique genomic barcode in the sample. Determine the mean false-positive rate from the negative control reactions and subtract this value from the values obtained in experimental reactions. Multiply values as necessary based on the dilutions that were performed when setting up each experiment (Table 3).
9. Determine Relative Fitness of an Organism by Calculating the CI or COI from Digital PCR-based Quantification of Barcoded Strains
- Calculate the CI of a barcoded strain using the following formulas where: AOutput is the absolute quantification of the barcoded strain at a given timepoint, WTOutput is the absolute quantification of barcoded wild-type bacteria at the same timepoint, AInput is the absolute quantification of the input inoculum of the barcoded strain, WTInput is the absolute quantification of the input inoculum of barcoded wild-type bacteria, XOutput is the summation of all strains at the same timepoint, XInput is the summation of the total input inoculum of all barcoded strains.
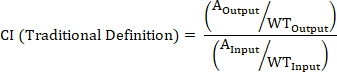
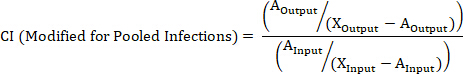
NOTE: See the discussion for advantages, disadvantages, and the most appropriate use of each formula.
- Repeat step 9.1 for each barcoded strain at all timepoints.
- If applicable, calculate the COI using the following formulas where: AOutput is the absolute quantification of a barcoded strain with mutated gene A at a given timepoint, ABOutput is the absolute quantification of a barcoded strain with mutated genes A and B at the same timepoint, AInput is the absolute quantification of the input inoculum of the barcoded strain with mutated gene A, ABInput is the absolute quantification of the input inoculum of barcoded strain with mutated genes A and B, BOutput is the absolute quantification of a barcoded strain with mutated gene B at a given timepoint, and BInput is the absolute quantification of the input inoculum of the barcoded strain with mutated gene B.
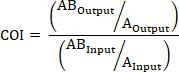
AND/OR
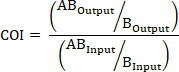
The use of this methodology requires that appropriate control reactions are performed to validate the sensitivity and specificity of each probe used to identify target DNA. In this representative experiment, we validated eight unique DNA barcodes with the eight corresponding probes for identification. All eight probes had a low rate of false positives in both NTC and negative control reactions (Table 3), highlighting their specificity even among highly similar DNA sequences. To assess the sensitivity of each condition, gDNA containing a unique barcode was serially diluted in a constant background of gDNA containing each of the seven remaining barcode sequences. With the approach outlined above, digital PCR could distinguish as few as 2 copies of gDNA in a background of nearly 2,000,000 similar DNA sequences (Table 3).
In addition to determining sensitivity and specificity of each probe and DNA barcode sequence, the dilutions performed in the validation study allowed us to calculate a simulated competitive index from the resulting data. While there was no true input or output for this experiment, the data can be analyzed as though a competition experiment has been performed. To do so, we consider each mixture in the serial dilution as an output (AOutput) for the diluted barcode, while the total output (XOutput), input (AInput), and the total input (XInput) of each strain is calculated from the quantification in the positive controls where all barcodes are included. Using the dilution factor for each mixture, the theoretical CI was determined and is reported in Table 3. In each of the dilution series that was performed for each barcode, the average simulated CI is reported along with the standard deviations for each duplicate dilution series. In all cases, the simulated CI that was calculated is similar to the theoretical CI. The majority of calculated CIs deviate from the theoretical CIs by less than 25%. In cases of lower theoretical CIs, the deviation of the calculated value was upwards of 2-fold. For example, this represented a change from a theoretical CI of 0.000625 to a calculated CI of 0.001220. These data highlight that the described method is both highly accurate and highly precise. The combination of high sensitivity, specificity, accuracy, and precision enable this system to reliably detect differences in fitness that may otherwise go unnoticed.
After validating that genomic barcodes could be accurately detected and quantified, we performed in vitro competition experiments (Table 4). The first competition experiment utilized eight wild-type S. Typhimurium strains that each contained a unique DNA barcode. Each strain was grown overnight, and the eight cultures were mixed together in equal amounts. 100 µL of this mixed inoculum was used to inoculate 4.9 mL of sterile LB broth and the resulting culture was incubated at 37 °C with constant agitation. gDNA was harvested from the inoculum to calculate the exact input of each strain. The growth of the culture was monitored by measuring the absorbance at 600 nm (OD600). At OD600 = 0.5 (logarithmic phase), a sample was collected from each culture and gDNA was harvested. The remaining culture was returned to 37 °C with constant agitation until 8 hours post-inoculation when a final sample was collected and gDNA harvested (stationary). Results were calculated using the CI formula modified for pooled infections. As expected, all wild-type strains had CI values nearly equal to 1 (Table 4). A similar competition experiment was performed using eight mutant S. Typhimurium strains that each had unique barcodes in addition to a single-, double-, or triple-transketolase deficiency18. As shown previously, the strains all grew similarly in LB broth, with only a slight lag observed in the triple-transketolase-deficient strain. However, when the growth of each strain was assessed by analyzing the CI, a much more profound defect was observed for the transketolase-deficient strain (CI was compared to growth curves in Shaw et al.18). Furthermore, this experiment allowed us to assign a quantifiable value to each strain’s growth characteristics instead of merely qualitatively describing the growth patterns. CIs for each strain were calculated using both the traditional formula where each strain was only compared to wild-type and the modified formula where all input strains were considered. While the changes were small, the CI of the triple-transketolase-deficient strain was artificially low in the traditional formula because it does not account for the other six competing strains that all exhibited near-wild-type fitness.
| Strains | Genotype | Source or reference |
| S. Typhimurium ATCC 14028s | wild-type | ATCC |
| TT22236 | LT2 Salmonella carrying pTP2223 | (27) |
| DH5α | F– φ80lacZΔM15 Δ(lacZYA-argF)U169 recA1 endA1 hsdR17(rK–, mK+) phoA supE44 λ– thi-1 gyrA96 relA1 | (28) |
| JAS18077 | putP::AA::FRT | This study |
| JAS18080 | putP::AD::FRT | This study |
| JAS18083 | putP::AG::FRT | This study |
| JAS18088 | putP::AL::FRT | This study |
| JAS18091 | putP::AO::FRT | This study |
| JAS18096 | putP::AT::FRT | This study |
| JAS18099 | putP::AW::FRT | This study |
| JAS18100 | putP::AX::FRT | This study |
| JAS18122 | ΔtktA::FRT putP::AD::FRT | This study |
| JAS18130 | ΔtktB::FRT putP::AL::FRT | This study |
| JAS18138 | ΔtktC::FRT putP::AT::FRT | This study |
| JAS18125 | ΔtktA::FRT ΔtktB::FRT putP::AG::FRT | This study |
| JAS18133 | ΔtktA::FRT ΔtktC::FRT putP::AO::FRT | This study |
| JAS18141 | ΔtktB::FRT ΔtktC::FRT putP::AW::FRT | This study |
| JAS18142 | ΔtktA::FRT ΔtktB::FRT ΔtktC::FRT putP::AX::FRT | This study |
| Plasmids | ||
| pKD13 | bla FRT ahp FRT PS1 PS4 oriR6K | (14) |
| pPCR Script Cam SK+ | ColE1 ori; CmR | Stratagene/Aligent |
| pTP2223 | Plac lam bet exo tetR | (16) |
| pCP20 | bla cat cI857 PRflp pSC101 oriTS | (29) |
| pSKAP | ColE1 ori; CmR; bla FRT ahp FRT | This study |
| pSKAP_AA | ColE1 ori; CmR; bla FRT ahp FRT; AA | This study |
| pSKAP_AD | ColE1 ori; CmR; bla FRT ahp FRT; AD | This study |
| pSKAP_AG | ColE1 ori; CmR; bla FRT ahp FRT; AG | This study |
| pSKAP_AL | ColE1 ori; CmR; bla FRT ahp FRT; AL | This study |
| pSKAP_AO | ColE1 ori; CmR; bla FRT ahp FRT; AO | This study |
| pSKAP_AT | ColE1 ori; CmR; bla FRT ahp FRT; AT | This study |
| pSKAP_AW | ColE1 ori; CmR; bla FRT ahp FRT; AW | This study |
| pSKAP_AX | ColE1 ori; CmR; bla FRT ahp FRT; AX | This study |
Table 1: Strains and plasmids used in this study.
| Name | Sequence (5' – 3')1,2,3 |
| pSKAP SDM AA – F | AGAAGTCTCCTGCTGGTGCTTGAGTCGATTGTGTAGGCTGGAGC |
| pSKAP SDM AA – R | ACTCAAGCACCAGCAGGAGACTTCTCTCAAGACGTGTAATGCTG |
| pSKAP SDM AD – F | AAGAGCACGGTGAGGTGATAGTAGGCGATTGTGTAGGCTGGAGC |
| pSKAP SDM AD – R | CCTACTATCACCTCACCGTGCTCTTCTCAAGACGTGTAATGCTG |
| pSKAP SDM AG – F | AGTAGTGTCCTGGAGGAGCATGTGACGATTGTGTAGGCTGGAGC |
| pSKAP SDM AG – R | TCACATGCTCCTCCAGGACACTACTCTCAAGACGTGTAATGCTG |
| pSKAP SDM AL – F | ACCACACATCGAAGGCACTAGCTCTCTCAAGACGTGTAATGCTG |
| pSKAP SDM AL – R | AGAGCTAGTGCCTTCGATGTGTGGTCGATTGTGTAGGCTGGAGC |
| pSKAP SDM AO – F | GTCCACAACCACACTCAGTGATACTCTCAAGACGTGTAATGCTG |
| pSKAP SDM AO – R | AGTATCACTGAGTGTGGTTGTGGACCGATTGTGTAGGCTGGAGC |
| pSKAP SDM AT – F | ACCAGTGTCCGTGACATGGCTAGACCGATTGTGTAGGCTGGAGC |
| pSKAP SDM AT – R | GTCTAGCCATGTCACGGACACTGGTCTCAAGACGTGTAATGCTG |
| pSKAP SDM AW – F | ACGACTGAGTGATGTGGATGTGACGCGATTGTGTAGGCTGGAGC |
| pSKAP SDM AW – R | CGTCACATCCACATCACTCAGTCGTCTCAAGACGTGTAATGCTG |
| pSKAP SDM AX – F | ACTATCGTGGTGTAACGACAGGCTGCGATTGTGTAGGCTGGAGC |
| pSKAP SDM AX – R | CAGCCTGTCGTTACACCACGATAGTCTCAAGACGTGTAATGCTG |
| M13 – F | GTAAAACGACGGCCAG |
| putP Recombination – F | TAGCGATGGGAGAGAGGACACGTTAATTATTCCATTTTAA TGCAGCATTACACGTC |
| putP Recombination – R | TACTGCGGGTATTAATGCTGAAAACATCCATAACCCATTG CCTGCAGTTCGAAGTTCC |
| qPCR Barcode Region Amplify – F1 | TGCAGCATTACACGTCTTG |
| qPCR Barcode Region Amplify – R2 | TAGGAACTTCGAAGCAGC |
| Barcode AA Probe – FAM | 6-FAM/AGAAGTCTC/ZEN/CTGCTGGTGCTTGAGTC/IBFQ |
| Barcode AD Probe – FAM | 6-FAM/AAGAGCACG/ZEN/GTGAGGTGATAGTAGGC/IBFQ |
| Barcode AG Probe – FAM | 6-FAM/AGTAGTGTC/ZEN/CTGGAGGAGCATGTGAC/IBFQ |
| Barcode AL Probe – FAM | 6-FAM/AGAGCTAGT/ZEN/GCCTTCGATGTGTGGTC/IBFQ |
| Barcode AO Probe – HEX | HEX/AGTATCACT/ZEN/GAGTGTGGTTGTGGACC/IBFQ |
| Barcode AT Probe – HEX | HEX/ACCAGTGTC/ZEN/CGTGACATGGCTAGACC/IBFQ |
| Barcode AW Probe – HEX | HEX/ACGACTGAG/ZEN/TGATGTGGATGTGACGC/IBFQ |
| Barcode AX Probe – HEX | HEX/ACTATCGTG/ZEN/GTGTAACGACAGGCTGC/IBFQ |
| 1Underlined nucleotides indicate complementary sequences for each DNA barcode that is inserted onto pSKAP after SDM. 2Double underlined nucleotides indicate a complementary region on the S. Typhimurium chromosome used for allelic replacement. 3PrimeTime qPCR Probes are hybridization oligos labelled with a 5' fluorescent dye, either 6-carboxyfluorescein (6-FAM) or hexachlorofluorescein (HEX), an internal quencher (ZEN), and the 3' quencer Iowa Black® FQ (IBFQ). |
|
Table 2: Primers and probes used in this study.
| Quantification (copies/20µL reaction) | ||||||||
| Description | AA | AD | AG | AL | AO | AT | AW | AX |
| NTC | N/A | 0.000 | 0.000 | 3.510 | N/A | 0.000 | 0.000 | 0.000 |
| NTC | 3.600 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| NTC | 3.510 | 0.000 | 1.280 | 1.180 | 2.340 | 0.000 | 0.000 | 0.000 |
| NTC | 2.290 | 0.000 | 0.000 | 1.200 | 1.150 | 1.160 | 0.000 | 0.000 |
| Mean | 3.133 | 0.000 | 0.320 | 1.473 | 1.163 | 0.290 | 0.000 | 0.000 |
| Negative | 5.130 | 1.156 | 0.000 | 1.124 | 3.745 | 1.281 | 7.354 | 7.142 |
| Negative | 5.270 | 0.000 | 0.000 | 1.087 | 1.666 | 1.643 | 7.746 | 2.269 |
| Negative | 2.660 | 0.000 | 1.361 | 1.451 | 8.974 | 0.000 | N/A | 0.000 |
| Negative | 6.090 | 0.000 | 0.000 | 2.251 | 0.000 | 2.531 | 8.700 | 3.495 |
| Negative | 1.740 | 0.000 | 1.086 | 2.130 | 4.171 | 0.000 | 7.113 | 5.522 |
| Negative | 6.220 | 3.581 | 0.000 | 4.022 | 1.175 | 1.341 | N/A | 5.950 |
| Mean | 4.518 | 0.789 | 0.408 | 2.011 | 3.288 | 1.133 | 7.728 | 4.063 |
| Positive | 22281.540 | 42673.039 | 46442.242 | 45359.180 | 47885.625 | 15708.027 | 45325.906 | 20810.559 |
| Positive | 23989.676 | 44625.523 | 47356.438 | 45790.660 | 47456.973 | 15096.601 | 47929.840 | 22455.234 |
| Positive | 17846.824 | 38980.133 | 45633.809 | 44174.820 | 33875.039 | 15156.063 | 42536.270 | 21467.840 |
| Positive | 21047.588 | 40140.848 | 41672.648 | 46028.496 | 47426.527 | 16718.000 | 46978.664 | 19876.473 |
| Positive | 20218.238 | 44660.602 | 41718.707 | 45799.375 | 46495.602 | 14590.264 | 54741.023 | 22011.938 |
| Positive | 18531.740 | 41620.801 | N/A | 48082.313 | 35645.199 | 15341.382 | 48950.992 | 21117.559 |
| Mean | 20652.601 | 42116.824 | 44564.769 | 45872.474 | 43130.827 | 15435.056 | 47743.783 | 21289.934 |
| Blank Subtraction | 20648.083 | 42116.035 | 44564.361 | 45870.463 | 43127.539 | 15433.923 | 47736.054 | 21285.871 |
| Undiluted A | 23024.961 | 44448.875 | 58897.510 | 51120.948 | 55450.191 | 18155.305 | 62844.068 | 27567.828 |
| Undiluted B | 18278.174 | 35252.586 | 54409.510 | 66022.396 | 43101.148 | 15732.609 | 60761.328 | 26581.979 |
| 1/50 A | 521.670 | 755.035 | 1066.898 | 1287.187 | 1053.339 | 181.324 | 1278.336 | 580.961 |
| 1/50 B | 435.326 | 634.215 | 1168.087 | 1383.537 | 991.040 | 165.443 | 1180.445 | 596.461 |
| 1/100 A | 228.028 | 598.848 | 603.911 | 631.116 | 507.956 | 258.405 | 665.647 | 331.590 |
| 1/100 B | 256.330 | 585.834 | 583.325 | 670.875 | 459.325 | 289.207 | 638.916 | 307.948 |
| 1/200 A | 121.283 | 305.293 | 258.247 | 346.965 | 234.774 | 114.163 | 169.055 | 172.553 |
| 1/200 B | 114.638 | 313.040 | 253.685 | 297.216 | 191.637 | 179.895 | 280.989 | 147.297 |
| 1/400 A | 42.829 | 141.343 | 127.337 | 163.605 | N/A | 71.241 | 157.697 | 85.976 |
| 1/400 B | 59.544 | 180.543 | 162.080 | 162.108 | 115.508 | 104.682 | 151.141 | 87.804 |
| 1/800 A | 34.304 | 67.934 | 65.939 | 83.857 | 66.134 | 31.784 | 82.722 | 45.616 |
| 1/800 B | 20.390 | 80.222 | 81.453 | 85.325 | 53.102 | 38.034 | 55.460 | 29.660 |
| 1/1600 A | 15.405 | 44.505 | 47.672 | 39.613 | 33.027 | 18.006 | 37.655 | 20.988 |
| 1/1600 B | 22.091 | 48.828 | 46.781 | 37.388 | 30.245 | 30.138 | 34.553 | 19.795 |
| 1/3200 A | 12.333 | 22.104 | 16.850 | 18.403 | 11.460 | 10.535 | 21.245 | 9.218 |
| 1/3200 B | 6.796 | 32.555 | 15.742 | 26.249 | 15.111 | 9.908 | 23.393 | 10.175 |
| Blank Subtraction | ||||||||
| Undiluted A | 23020.443 | 44448.086 | 58897.103 | 51118.937 | 55446.903 | 18154.172 | 62836.339 | 27563.765 |
| Undiluted B | 18273.655 | 35251.797 | 54409.103 | 66020.385 | 43097.860 | 15731.477 | 60753.600 | 26577.916 |
| 1/50 A | 517.152 | 754.246 | 1066.490 | 1285.176 | 1050.051 | 180.192 | 1270.607 | 576.898 |
| 1/50 B | 430.808 | 633.426 | 1167.679 | 1381.526 | 987.751 | 164.311 | 1172.717 | 592.398 |
| 1/100 A | 223.509 | 598.059 | 603.503 | 629.105 | 504.668 | 257.272 | 657.918 | 327.527 |
| 1/100 B | 251.812 | 585.044 | 582.918 | 668.864 | 456.036 | 288.074 | 631.187 | 303.885 |
| 1/200 A | 116.765 | 304.503 | 257.840 | 344.954 | 231.486 | 113.030 | 161.327 | 168.490 |
| 1/200 B | 110.120 | 312.251 | 253.277 | 295.205 | 188.348 | 178.762 | 273.261 | 143.234 |
| 1/400 A | 38.310 | 140.554 | 126.929 | 161.594 | N/A | 70.108 | 149.968 | 81.913 |
| 1/400 B | 55.026 | 179.753 | 161.672 | 160.097 | 112.219 | 103.549 | 143.413 | 83.741 |
| 1/800 A | 29.786 | 67.145 | 65.531 | 81.847 | 62.846 | 30.651 | 74.994 | 41.553 |
| 1/800 B | 15.872 | 79.433 | 81.045 | 83.314 | 49.813 | 36.901 | 47.732 | 25.596 |
| 1/1600 A | 10.886 | 43.716 | 47.264 | 37.602 | 29.739 | 16.874 | 29.927 | 16.925 |
| 1/1600 B | 17.573 | 48.039 | 46.373 | 35.377 | 26.957 | 29.005 | 26.825 | 15.732 |
| 1/3200 A | 7.815 | 21.314 | 16.442 | 16.392 | 8.172 | 9.402 | 13.517 | 5.155 |
| 1/3200 B | 2.278 | 31.765 | 15.334 | 24.238 | 11.822 | 8.776 | 15.664 | 6.112 |
| Simulated CI | ||||||||
| Undiluted A | 1.114895 | 1.055372 | 1.321619 | 1.114419 | 1.285650 | 1.176251 | 1.316329 | 1.294932 |
| Undiluted B | 0.885005 | 0.837016 | 1.220911 | 1.439279 | 0.999312 | 1.019279 | 1.272698 | 1.248618 |
| 1/50 A | 0.025046 | 0.017909 | 0.023931 | 0.028018 | 0.024348 | 0.011675 | 0.026617 | 0.027102 |
| 1/50 B | 0.020864 | 0.015040 | 0.026202 | 0.030118 | 0.022903 | 0.010646 | 0.024567 | 0.027831 |
| 1/100 A | 0.010825 | 0.014200 | 0.013542 | 0.013715 | 0.011702 | 0.016669 | 0.013782 | 0.015387 |
| 1/100 B | 0.012195 | 0.013891 | 0.013080 | 0.014582 | 0.010574 | 0.018665 | 0.013222 | 0.014276 |
| 1/200 A | 0.005655 | 0.007230 | 0.005786 | 0.007520 | 0.005367 | 0.007323 | 0.003380 | 0.007916 |
| 1/200 B | 0.005333 | 0.007414 | 0.005683 | 0.006436 | 0.004367 | 0.011582 | 0.005724 | 0.006729 |
| 1/400 A | 0.001855 | 0.003337 | 0.002848 | 0.003523 | N/A | 0.004542 | 0.003142 | 0.003848 |
| 1/400 B | 0.002665 | 0.004268 | 0.003628 | 0.003490 | 0.002602 | 0.006709 | 0.003004 | 0.003934 |
| 1/800 A | 0.001443 | 0.001594 | 0.001470 | 0.001784 | 0.001457 | 0.001986 | 0.001571 | 0.001952 |
| 1/800 B | 0.000769 | 0.001886 | 0.001819 | 0.001816 | 0.001155 | 0.002391 | 0.001000 | 0.001203 |
| 1/1,600 A | 0.000527 | 0.001038 | 0.001061 | 0.000820 | 0.000690 | 0.001093 | 0.000627 | 0.000795 |
| 1/1,600 B | 0.000851 | 0.001141 | 0.001041 | 0.000771 | 0.000625 | 0.001879 | 0.000562 | 0.000739 |
| 1/3,200 A | 0.000378 | 0.000506 | 0.000369 | 0.000357 | 0.000189 | 0.000609 | 0.000283 | 0.000242 |
| 1/3,200 B | 0.000110 | 0.000754 | 0.000344 | 0.000528 | 0.000274 | 0.000569 | 0.000328 | 0.000287 |
| Average CI (Theoretical) | ||||||||
| Undiluted (1) | 0.999950 | 0.946194 | 1.271265 | 1.276849 | 1.142481 | 1.097765 | 1.294514 | 1.271775 |
| 1/50 (0.02) | 0.022955 | 0.016474 | 0.025067 | 0.029068 | 0.023625 | 0.011161 | 0.025592 | 0.027466 |
| 1/100 (0.01) | 0.011510 | 0.014046 | 0.013311 | 0.014148 | 0.011138 | 0.017667 | 0.013502 | 0.014832 |
| 1/200 (0.005) | 0.005494 | 0.007322 | 0.005735 | 0.006978 | 0.004867 | 0.009453 | 0.004552 | 0.007322 |
| 1/400 (0.0025) | 0.002260 | 0.003803 | 0.003238 | 0.003507 | 0.00260* | 0.005626 | 0.003073 | 0.003891 |
| 1/800 (0.00125) | 0.001106 | 0.001740 | 0.001645 | 0.001800 | 0.001306 | 0.002188 | 0.001285 | 0.001577 |
| 1/1,600 (0.000625) | 0.000689 | 0.001089 | 0.001051 | 0.000795 | 0.000657 | 0.001486 | 0.000594 | 0.000767 |
| 1/3,200 (0.000313) | 0.000244 | 0.000630 | 0.000357 | 0.000443 | 0.000232 | 0.000589 | 0.000306 | 0.000265 |
| Standard Deviation | ||||||||
| Undiluted | 0.11494 | 0.10918 | 0.05035 | 0.16243 | 0.14317 | 0.07849 | 0.02182 | 0.02316 |
| 1/50 | 0.00209 | 0.00143 | 0.00114 | 0.00105 | 0.00072 | 0.00051 | 0.00103 | 0.00036 |
| 1/100 | 0.00069 | 0.00015 | 0.00023 | 0.00043 | 0.00056 | 0.00100 | 0.00028 | 0.00056 |
| 1/200 | 0.00016 | 0.00009 | 0.00005 | 0.00054 | 0.00050 | 0.00213 | 0.00117 | 0.00059 |
| 1/400 | 0.00040 | 0.00047 | 0.00039 | 0.00002 | 0* | 0.00108 | 0.00007 | 0.00004 |
| 1/800 | 0.00034 | 0.00015 | 0.00017 | 0.00002 | 0.00015 | 0.00020 | 0.00029 | 0.00037 |
| 1/1,600 | 0.00016 | 0.00005 | 0.00001 | 0.00002 | 0.00003 | 0.00039 | 0.00003 | 0.00003 |
| 1/3,200 | 0.00013 | 0.00012 | 0.00001 | 0.00009 | 0.00004 | 0.00002 | 0.00002 | 0.00002 |
| *Represents results from a single experiment. | ||||||||
Table 3: Absolute quantification and simulated CI calculation.
| Condition | Competitive Index1 | |||||||
| Experiment 1 | ||||||||
| WTAA | WTAD | WTAG | WTAL | WTAO | WTAT | WTAW | WTAX | |
| Logarithmic | 0.927 ± 0.033 | 0.992 ± 0.031 | 1.068 ± 0.025 | 0.921 ± 0.02 | 1.044 ± 0.03 | 1.051 ± 0.057 | 1.094 ± 0.027 | 0.929 ± 0.005 |
| Stationary | 1.1 ± 0.021 | 1.071 ± 0.053 | 1.079 ± 0.065 | 0.948 ± 0.02 | 0.98 ± 0.02 | 0.873 ± 0.044 | 0.97 ± 0.056 | 1.021 ± 0.007 |
| Experiment 2 | ||||||||
| CI (Traditional) | WTAA | ΔAAD | ΔBAL | ΔCAT | ΔABAG | ΔACAO | ΔBCAW | ΔABCAX |
| Logarithmic | 1 ± 0 | 0.802 ± 0.084 | 0.957 ± 0.02 | 0.989 ± 0.073 | 0.581 ± 0.153 | 0.86 ± 0.053 | 0.995 ± 0.011 | 0.695 ± 0.061 |
| Stationary | 1 ± 0 | 0.97 ± 0.063 | 1.043 ± 0.058 | 0.99 ± 0.036 | 1.625 ± 0.589 | 0.835 ± 0.051 | 0.912 ± 0.047 | 0.477 ± 0.049 |
| CI (Pooled Inoculum) | ||||||||
| Logarithmic | 1.114 ± 0.039 | 0.864 ± 0.074 | 1.073 ± 0.032 | 1.1 ± 0.068 | 0.633 ± 0.152 | 0.938 ± 0.056 | 1.111 ± 0.043 | 0.746 ± 0.06 |
| Stationary | 1.078 ± 0.039 | 1.039 ± 0.049 | 1.166 ± 0.093 | 1.066 ± 0.01 | 1.735 ± 0.613 | 0.876 ± 0.035 | 0.97 ± 0.045 | 0.49 ± 0.047 |
| 1Values represent mean CI ± standard deviation for three or four replicate experiments. | ||||||||
Table 4: Representative results from in vitro competition between S. Typhimurium strains.
Table S1. Optional primers for creating additional barcode sequences and corresponding fluorescent probes for their detection. Please click here to download this file.
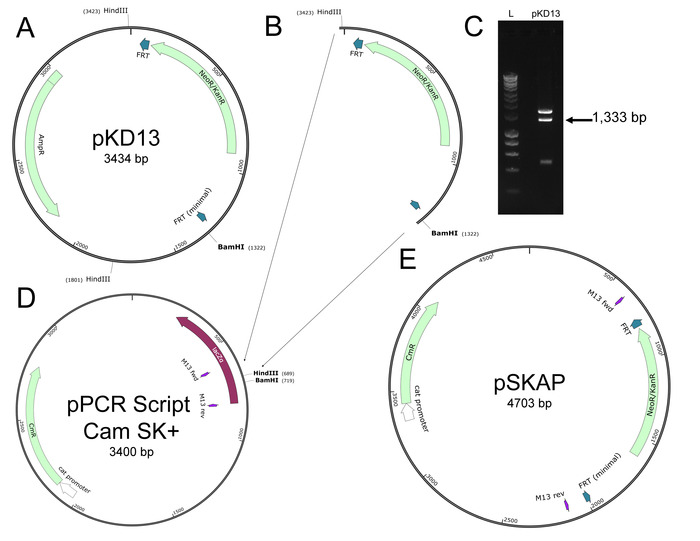
Figure 1. Generation of pSKAP. (A) Purified pKD13 was subjected to restriction digestion with HindIII and BamHI. (B, C) The 1,333 bp fragment of interest containing an FRT-flanked kanamycin resistance gene was purified. (D) pPCR Script Cam SK+ was also digested with HindIII and BamHI and the fragment from pKD13 (B) was ligated in to generate (E) pSKAP. Please click here to view a larger version of this figure.

Figure 2: Insertional site-directed mutagenesis to pSKAP. (A) Insertion of 25 bp DNA barcodes at position 725 was performed using PCR. Forward and reverse primers specific to that location were designed with complementary 25-nucleotide 5’ extensions (denoted in the primer as lowercase “a”). (B) A generic pSKAP_Barcode plasmid resulting from SDM is shown with the location of the inserted DNA barcode highlighted orange. Please click here to view a larger version of this figure.
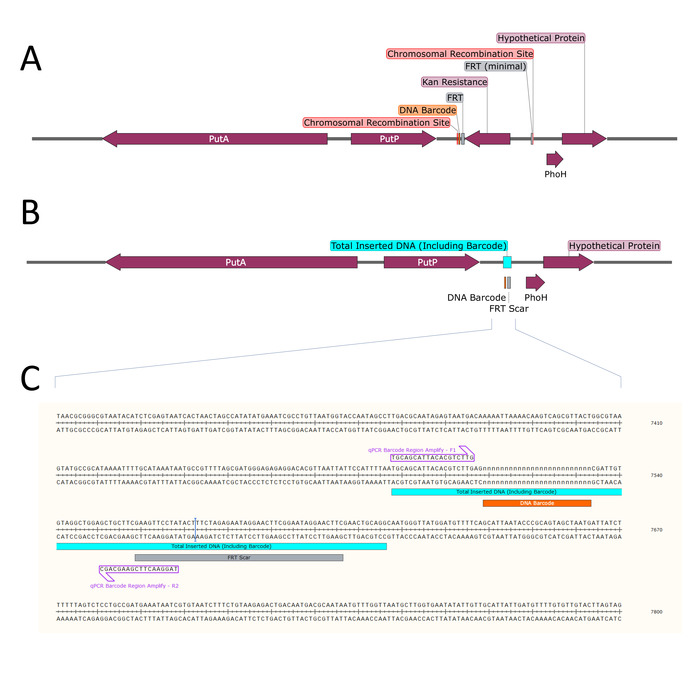
Figure 3: Chromosomal rearrangement downstream of putP. (A) After λ-Red mediated recombination, the selectable kanamycin resistance gene (dark purple) flanked by FRT sites (grey) is inserted on the chromosome between the loci indicated (Chromosomal Recombination Site, red). The unique DNA barcode (orange) is inserted just outside the FRT site. (B) The kanamycin resistance gene is removed by FRT-mediated excision, leaving a remnant of inserted DNA on the chromosome (Total Inserted DNA, blue) consisting of the DNA barcode and an FRT scar. (C) The modified chromosomal DNA sequence surrounding the inserted DNA is shown, along with the amplification priming sites (light purple) used for digital PCR. Please click here to view a larger version of this figure.

Figure 4: Dilution scheme for validating fluorescent probe sensitivity and specificity. (A) Purified gDNA from seven barcoded strains is mixed together in equal amounts to create the diluent for diluting the omitted barcoded gDNA (AX in the example above). (B) Perform a serial dilution of the omitted barcoded gDNA (AX in the example above) using the prepared diluent described previously. Thoroughly mix the contents of each tube before transferring to the next tube. Figure 4 was created with BioRender. Please click here to view a larger version of this figure.
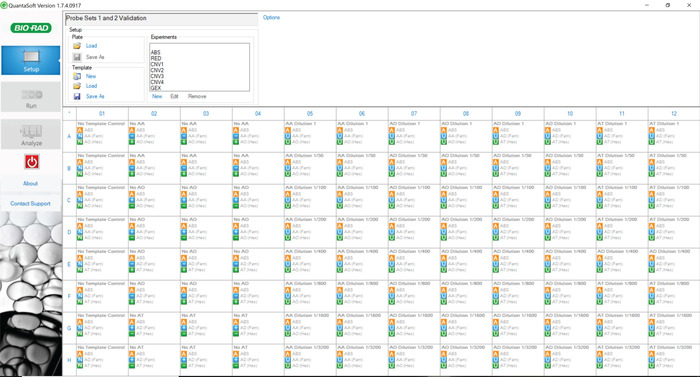
Figure 5: Plate layout for analyzing sensitivity and specificity of primer-probe sets 1 and 2. The digital PCR experiment includes NTCs, positive controls, negative controls, and the dilution schemes for each of the tested barcodes. The plate for validating primer-probe sets 3 and 4 is laid out in the same pattern using the appropriate barcoded gDNA. Please click here to view a larger version of this figure.
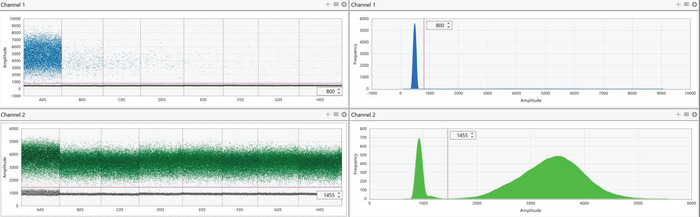
Figure 6: Representative digital PCR results of diluted AA-barcoded gDNA. gDNA containing the AA barcode was diluted in a background of all other barcoded gDNA as described in Figure 4. Channel 1 represents the FAM probe for the AA barcode (top panels) while channel 2 represents the HEX probe for the AO barcode (bottom panels). Results of each probe are presented as both individual droplet fluorescent amplitude (left panels) and a histogram representing the frequency of fluorescent intensity of all droplets in the selected wells (right panels). For each condition, positive (high fluorescence) and negative (low fluorescence) droplets should form two distinct populations. In the case of AA that was diluted, and most droplets were negative, the histogram (top right panel) appears to only depict a single population. This is because positive droplets are substantially outnumbered by negative droplets; however, two distinct populations are still visible by examining droplet fluorescent amplitude in the left panels. The populations should be separated using the threshold feature to define positive and negative droplets (visualized by the pink line). Threshold values will vary depending on the probes that were used, but all wells that utilize the same probe mix should have identical thresholds. As the AA-barcoded gDNA was diluted, there is a decrease in positive (high fluorescent) droplets while the number of positive AO droplets remains constant in the background. Please click here to view a larger version of this figure.
