CAUTION: For the entirety of the protocol, it is advisable to conduct all wet-lab procedures sterilely by use of lab coats, disposable gloves and sterile reagents and equipment. It is also advisable to prepare PCR reactions in a separate room (pre-PCR) not used for PCR amplification and/or handling of PCR products (post-PCR). Store all reagents as recommended by the manufacturer. See Table of Materials for further details on reagents, equipment and software used.
1. Bacterial Cultivation and Extraction of Genomic DNA
- Sow out Y. ruckeri pure cultures on any suitable agar type (the authors used 5% bovine blood agar) and incubate at 22 °C for 1-2 days, or 15 °C for 3-4 days.
- From each agar plate, pick a single representative colony with an inoculation loop and transfer to 1.5 mL centrifuge tubes containing 50 µL of ultrapurified water. Suspend, vortex briefly, and incubate for 7 min on a heating block at 100 °C.
- Centrifuge at 16 000 x g for 3 min and use a pipette to carefully transfer the supernatant into an empty 1.5 mL centrifuge tube. Proceed to next step using the supernatant as template DNA or store at -20 °C until such time.
2. Multiplex PCR setup and Cycling Conditions
NOTE: Each multiplex PCR reaction (two per Y. ruckeri isolate) should contain 12.5 µL of 2x Multiplex PCR Plus master mix, 0.1 to 0.2 µM of each appropriate primer pair (Table 1) and 3 µL of template DNA, adjusted to a final reaction volume of 25 µL by addition of RNase-free water. Aim to keep light exposure of the fluorescently labelled forward-primers at a minimum (e.g., by wrapping their storage tubes in aluminum foil).
- For each of the two multiplex PCR assays (Table 1), prepare master mixes as described above (without template DNA) according to the number of samples plus one positive and one negative control. Additionally, allow 10% surplus volume. Vortex the prepared master mixes gently at low speed.
- Distribute 22 µL of each master mix separately into individual wells on either PCR strips or plates, as appropriate for the number of samples, and add 3 µL of template to each well (for positive and negative controls, respectively, use DNA from a verified Y. ruckeri strain and ultrapurified water). Seal and centrifuge briefly.
- Run all samples on a PCR thermal cycler with the following programme: (i) 5 min at 95 °C (ii) 30 cycles of 0.5 min at 95 °C, 1.5 min at 60 °C, and 1 min at 72 °C, and (iii) 60 min at 68 °C, followed by cooling to 4 °C indefinitely. The program will complete in less than 3 h.
3. PCR Amplicon Confirmation by Gel Electrophoresis
- According to the manufacturer’s recommendations, prepare a volume of 1.5% (w/v) agarose gel in 1x tris-borate-EDTA (TBE) buffer appropriate for the number of PCR reactions to be tested. Prior to casting, add 5 µL of fluorescent nucleic acid dye per 50 µL of gel solution and mix. Use trays and combs as appropriate for casting, leaving an appropriate number of wells free for DNA reference ladders.
- After setting, submerge the gel in 1x TBE-buffer in a GE system. Mix 5 µL of PCR product together with 2 µL of loading dye and transfer to gel wells. Add 5 µL of DNA ladder in empty wells for reference.
- Run the gel at 110 V per 15 cm for approximately 1 h and use a UV-based gel imaging/visualisation system to verify the presence of multiple (up to five) bands representing PCR amplicons (see example in Figure 1). Discard the gel. Proceed to the next step or store remaining PCR products at 4 °C until further processing.
4. Capillary Electrophoresis Setup and Run Conditions
- Following confirmation of PCR amplicons, dilute PCR products 1:10 (v/v) in purified water. Seal, mix and centrifuge briefly.
- Working in a fume cupboard, prepare a volume of master mix consisting of 9 µL of formamide and 0.5 µL of size standard per PCR product (allow 10% surplus volume). Vortex briefly and distribute 9.5 µL into wells on a plate appropriate for the available CE system, before adding 0.5 µL of diluted PCR product. Seal, mix and centrifuge briefly.
CAUTION: Handle with care. Mixing formamide with water generates formic acid, which is toxic. - Using a PCR thermal cycler, denature the samples at 95 °C for 3 min before cooling to 4 °C indefinitely. Centrifuge briefly and load the plate onto a calibrated CE system according to the manufacturer’s instructions.
- Run fragment analysis CE using reagents as appropriate for the apparatus of choice and the following settings: 60 °C; 5 s injections at 1.6 kV (32 V per cm); 32 min run time at 15 kV (300 V per cm). CE fragment analysis of 24 wells on a 24-capillary (50 cm) will typically take approximately 50 min.
5. VNTR Size Calling, Repeat Count Calculation and MLVA Profiling
NOTE: Step 5.1 describes Y. ruckeri VNTR CE size calling from electropherogram files, using the specific software listed in Table of Materials. Consult the software manual for additional details and troubleshooting. For use of other software, consult appropriate manuals.
- Import CE result files (two per Y. ruckeri isolate). Set Analysis Method to Microsatellite Default and select the appropriate product choice under Size Standard, prior to pressing the Analyze button. Verify the correct identification of size standard fragments through the Size Match Editor and rectify any visibly erroneous allocations.
- Having selected the sample(s) to be read, hit the Display Plots button and press Ctrl+A to enable view of the Sizing Table. While in the top panel, hold down Ctrl while clicking on the five peaks representing the VNTR amplicons (use zooming tool as needed).
NOTE: For each of the multiplex PCR products, the electropherogram will show five peaks distributed amongst the three dyes employed (see 5' dye labelling of forward primers in Table 1 and the two examples in Figure 2). - Press Ctrl+G to filter the Sizing Table, showing only characteristics of the five highlighted peaks, and record CE size calls for each VNTR locus (with reference to Table 1) for downstream application.
- Having selected the sample(s) to be read, hit the Display Plots button and press Ctrl+A to enable view of the Sizing Table. While in the top panel, hold down Ctrl while clicking on the five peaks representing the VNTR amplicons (use zooming tool as needed).
- In order to account for biased amplicon mobility patterns during CE, calculate accurate VNTR repeat counts according to the formula provided below, employing VNTR CE size calls together with locus-specific variables (see Table 1). For efficiency, it is advisable to automate this process (e.g., by using a spreadsheet template).
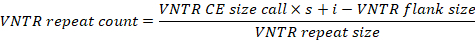
- Round calculated VNTR repeat counts off to the nearest integer and concatenate into ten-digit strings, each representing the MLVA profile of a single Y. ruckeri isolate.
6. Minimum Spanning Tree Cluster Analysis of MLVA Data
NOTE: Step 6 describes the creation of MST diagrams from Y. ruckeri MLVA data, using the specific software listed in Table of Materials. Consult the software manual for additional details and troubleshooting. For use of other software, consult appropriate manuals.
- Create a new database and opt to activate the MLVA plugin.
- Import Y. ruckeri MLVA profiles and metadata by selecting Character type data followed by Import fields and characters (further sub-selection depending on storage format). When prompted, specify import rules according to the content of the import file: In the Destination type column, classify VNTR repeat counts as Character value: VNTR, and the miscellaneous metadata as Entry information: Entry info field.
NOTE: For comparison and context, it is also possible to import the entire dataset published (open access) together with the original paper employing the present MLVA protocol14. MLVA profiles and metadata on the diverse collection of Y. ruckeri isolates (n = 484) scrutinised in that study is available from its supplemental material (Tables S1 and S2) through the following link: https://aem.asm.org/content/84/16/e00730-18/figures-only#fig-data-additional-files - In the Experiment type panel, open the VNTR entry and set minimum and maximum values for each VNTR locus to 0 and 100, respectively. Under General settings, set the number of decimal digits to 0 and select Numbers under Data type. Check to consider absent values as zero.
- Select imported samples destined for MST cluster analysis and click the Create new comparison button (in Comparison panel).
- If desired for the visual presentation of the MST, allocate the samples to colored groups (e.g., according to a particular metadata trait) by employing the various options available in the Groups panel.
NOTE: Groups can also be created/altered retrospectively, subsequent to the following steps. - Select Advanced cluster analysis… and MST for categorical data to generate an MST diagram based on the chosen samples.
- Further modify the visual presentation of the MST as preferred (e.g., by adding partitioning parameters, node/branch labelling, crosslinks, legends, etc). See example in Figure 3.
NOTE: A cluster (clonal complex) partitioning threshold of ≤4/10 non-identical VNTR loci, in addition to hiding of branch connections representing >5/10 non-identical VNTR loci, has previously been employed for MST cluster analysis based on MLVA data generated using this protocol14. Provided the aforementioned dataset of 484 Y. ruckeri MLVA profiles was imported, those samples can also be included for MST cluster analysis (as described above) to provide a global and historical context. This will e.g. facilitate identification of any samples affiliated with previously described clonal complexes, as well as those representing yet undescribed lineages. Depending on available metadata, the resulting MST diagram can be scrutinised in different ways, e.g. to discover eventual clustering patterns linked to particular traits (geography, host, time etc.). - If needed, export the finalized MST in a desired format using the Export image selection.
- If desired for the visual presentation of the MST, allocate the samples to colored groups (e.g., according to a particular metadata trait) by employing the various options available in the Groups panel.
Following multiplex PCR as described here, a typical GE image verifying the presence of multiple amplicons from each PCR reaction is shown in Figure 1. Downstream CE fragment analysis performed on verified PCR products will, for each Y. ruckeri isolate examined, result in two electropherogram files used for size calling of the respective VNTR loci (Figure 2). From analysis of 484 diverse Y. ruckeri isolates, no overlap in amplicon size range was observed between VNTR loci labelled with the same dye in the same multiplex reaction (Table 1)14. Each of the electrophoretic peaks can, therefore, be unambiguously identified by color.
Following import of MLVA profiles and relevant metadata into the preferred software, MST diagrams can be constructed as described for scrutiny of any epidemiological patterns of interest in the material. Consult appropriate manuals for additional options available in the respective software. As an example, Figure 3 shows comparison by MST of MLVA profiles for Y. ruckeri isolates recovered from fish associated with five different salmon farms in Norway.
The consistent repeat sizes of the ten VNTR loci, as well as their in vitro and in vivo stability, have previously been verified in the original study based upon this protocol14. Briefly, this was done using Sanger sequencing (repeat size), and by MLVA typing of multiple isolates following serial passages (in vitro) and from within individual disease outbreaks (in vivo). Moreover, the environmental stability of the loci over time was examined by typing multiple 'house strain' isolates recovered over several years from persistently infected freshwater production sites for Atlantic salmon.
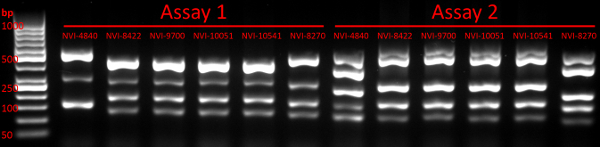
Figure 1: Gel electrophoresis verifying the presence of multiple PCR products. The image confirms the presence of multiple PCR amplicons in all 12 lanes containing samples, with the first lane representing the DNA ladder used. The sizes of selected ladder fragments have been indicated, as have the PCR assay and strain (see Table S1 in Gulla et al. 201814) affiliation of each lane. Please click here to view a larger version of this figure.
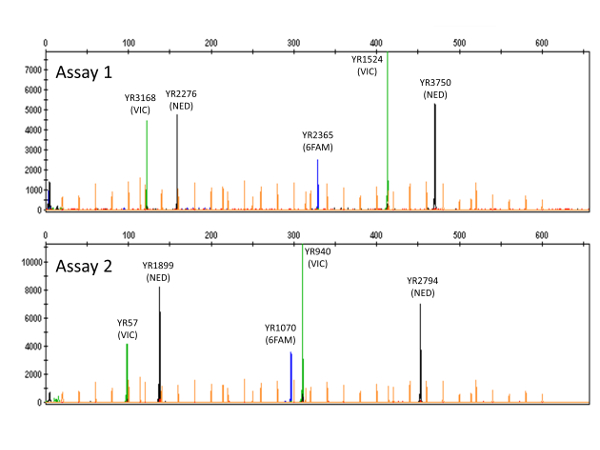
Figure 2: Electropherograms showing peaks corresponding to VNTR amplicons. Names of the different VNTR loci are indicated, with dye labels (VIC = green; NED = black; 6FAM = blue) in parentheses. The two electropherograms (PCR assay 1 top; PCR assay 2 bottom) originate from typing of a single Y. ruckeri isolate. Orange peaks (dye LIZ) represent the size standard employed. Please click here to view a larger version of this figure.
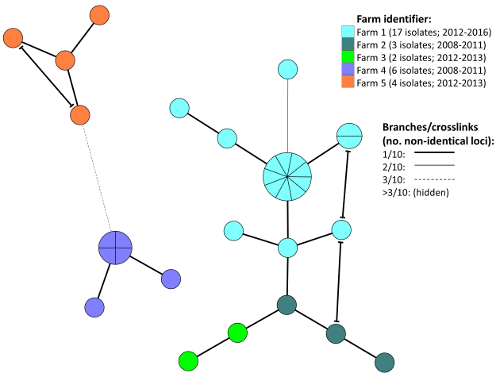
Figure 3: Example Minimum spanning tree for epidemiological evaluation. The diagram is based on MLVA profiles from Y. ruckeri isolates recovered from Atlantic salmon in five different Norwegian farms (1-5; see legend) experiencing recurrent yerisniosis outbreaks. A clear clustering tendency linked to farm origin can be observed. Crosslinks show all possible connections involving ≤1/10 non-identical VNTR loci (see legend). Please click here to view a larger version of this figure.
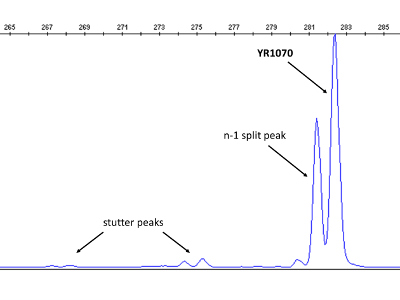
Figure 4: Electropherogram visualizing stutter and split peaks. In this case, both occur simultaneously, which is not always the case. The longer and taller peak, representing the YR1070 VNTR locus, can be readily distinguished. The display is magnified and shows only blue dye peaks. Please click here to view a larger version of this figure.
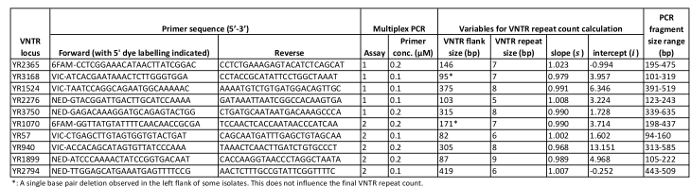
Table 1: VNTR locus characteristics. Relevant characteristics of the ten Y. ruckeri VNTR regions targeted in the present MLVA protocol.
