4sU labeling: Verify optimal 4sU-labeling conditions (apoptosis, nuclear stress, cytoplasmic stress), time, and concentration: High levels of 4sU can inhibit the production and processing of rRNA and induce cytoplasmic as well as nuclear stress30. Therefore, cells of interest should be tested for 4sU-induced stress as well as apoptosis. Western blot analysis is recommended for visualizing p53 accumulation, which indicates nuclear stress, increasing phospho-EIF2a levels which display cytoplasmic stress and fluorescence-activated cell sorting (FACS) analysis for apoptosis. High levels and lengthy exposure to 4sU or drugs like thapsigargin or arsenite can be used to induce cellular stress. To induce apoptosis or cell death, cells were treated with BH3I-1 (500 ng/µL) or incubated for 5 min at 95 °C (heat shock). Annexin V/7-AAD staining was used to determine apoptotic (Annexin V) and dead (7-AAD) cells. Labeling of in vitro generated primary Th1 cells for 0.5 h with 500 µM 4sU (final concentration) or 1 h with 200 µM 4sU neither induces signs of cellular stress nor apoptosis (Figure 1) but lead to sufficient 4sU incorporation.
RNA labeling time can also be shortened (≤5 min) which leads to an increase in short-lived intronic sequences compared to longer labeling times. To visualize co-transcriptional splicing rates, 4sU-labeling times should not exceed 30 min. For further details regarding 4sUlabeling, please refer to Rädle et al.19
Quality Control: RNA integrity is of great importance when processing RNA. It is most convenient to check the RNA quality of 4sU-labeled RNA after biotinylation by electrophoretical analysis (see Table of Materials). Consider verifying isolated RNA from step 2.2.2, especially when using it for sequencing of total RNA. RNA integrity number (RIN) should be ≥8 to ensure RNA integrity for further processing (Figure 3).
Electrophoretical analysis can also be used to verify newly transcribed RNA. Be aware that newly transcribed RNA contains significantly less mature rRNAs compared to total RNA with the typical rRNA bands being much less prominent.
Ribosome profiling: PCR amplification of the cDNA library: cDNA amplification (step 4.9.1) is a critical step to ensure good sequencing results. Analyze amplified libraries by electrophoretical analysis. A good sample of amplified libraries shows a peak around 140 – 160 bp (Figure 4A). Excessive amounts of adapter dimers should be avoided (Figure 4B) and those samples should be further purified using the PAGE purification procedure according to the manufacturer's protocol (PAGE purification of PCR products). Too much template or too many PCR cycles can result in over-amplification characterized by the appearance of higher-than-expected molecular weight bands, smeared PCR products, and adapter dimer products (Figure 4C). For most samples 1 – 5 µL of circularized cDNA and 9 PCR cycles for amplification will typically yield sufficient amounts of the correct PCR product.
ChIP: Chromatin shearing: Optimal shearing conditions need to be adjusted for each type of cell. Determine shearing conditions (e.g., number of cycles, high or low power) in advance. Use the same number of cells and the same volume for testing purposes, since a lower cell density increases the shearing efficiency. Try to avoid over- or under-shearing the chromatin. Large chromatin fragments can dramatically affect ChIP results by clogging, and over-shearing can destroy epitopes on the protein of interest, leading to a lower binding efficiency by the antibody. In this experiment, the best results were achieved when the main fraction of the sheared chromatin was around 1,000 bp or slightly lower (Figure 5A).
Verifying ChIP by qPCR: Before starting the ChIP, it is advisable to test if the used antibody is suitable for ChIP (if possible, use ChIP grade antibodies) by ChIP-qPCR. Verify the ChIP for sequencing by qPCR before starting the library preparation (see step 5.6.5). Design primers that bind to a known target site of the protein of interest. If the exact target site within a gene is unknown, several primer pairs can be used to scan the gene and associated regulatory elements. For RNAPII ChIP of Th1 cells Ifng, which is transcriptionally upregulated upon stimulation, and actin primers can be used as a positive control. Sox9 and insulin serve as a negative control, since these genes are not expressed in Th1 cells (Figure 5B). Remember not to use exon-spanning primers, which are normally used for qPCR of mRNA. An IgG control can also be used to prove specificity of the used antibody. Immunoprecipitated DNA can be measured with a suitable fluorometer (see Table of Materials). Amounts of nonspecifically bound DNA by the IgG control should be significantly lower compared to the DNA amount bound by the antibody of interest.
Replicates: proof of biological significance: It is strongly advised to perform the kinetic experiment for all methods starting from the same pool of cells to ensure cells have the same identity for all untreated and treated samples (Figure 2). Nevertheless, it is recommended to take small aliquots of main time points for each method to compare samples to a biological replicate (e.g., by qPCR, FACS analysis). This allows for a rough estimation if the treatment for both replicates was reproducible and you could proceed with sequencing. Validation of the replicates should be performed by means of bioinformatical analysis. Reproducibility of results can be assessed in terms of correlation between FPKM values between replicates and visualized using scatter plots (Figure 6).
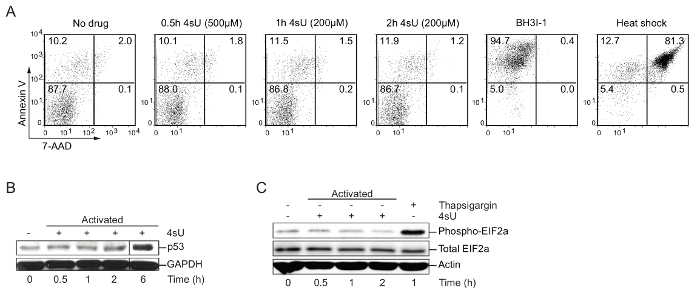
Figure 1: Verification of optimal 4sU-labeling conditions without perturbing cell physiology (Figure from Davari et al.11)
(A) Detection of cell apoptosis by FACS analysis: In vitro generated Th0 cells were treated with different concentrations of 4sU (indicated in brackets) for 0.5 h, 1 h, and 2 h, respectively. BH3I-1 treatment was used to induce apoptosis determined by Annexin V, whereas heat shock (5 min at 95 °C) was used to induce cell death determined by 7-AAD. (B) Western blot analysis for p53 of 4sU treated and activated T cells: samples were labeled with 200 µM 4sU for the indicated time of activation, except the 0.5 h time point that was labeled with 500 µM 4sU. (C) Western blot analysis of phospho-EIF2a and total EIF2a in activated Th1 cells with the same labeling conditions as in (B). Thapsigargin was used as a positive control. Please click here to view a larger version of this figure.
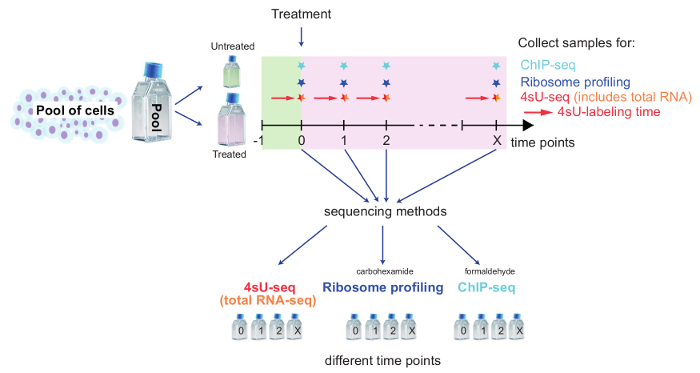
Figure 2: Schematic overview of a kinetic setup to track genome-wide changes
This scheme illustrates the setup for combining 4sU-seq, total RNA-seq, Ribosome Profiling, and ChIP-seq to study genome-wide changes upon cell treatment. Pool cells and set aside the required number of cells for untreated control. Treat remaining cells and split for each method and time point. Label untreated/treated cells for 4sU-seq with 4sU as described. Time points and samples for each method depend on the specific biological question being examined. Take samples for each time point and method, and follow the dedicated part of the protocol. Please click here to view a larger version of this figure.
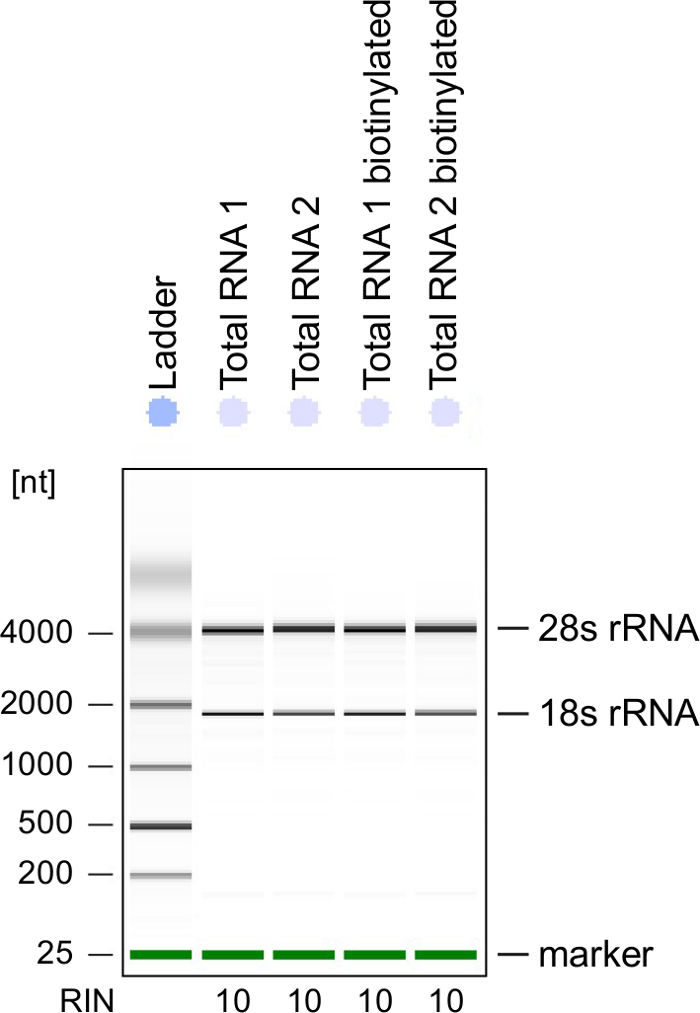
Figure 3: Quality control of 4sU-labeled RNA
Total RNA and biotinylated RNA obtained from activated Th1 cells were analyzed on a Bioanalyzer. 18S rRNA and 28S rRNA are shown and RNA integrity number (RIN) is given by the instrument to determine integrity of the RNA. RIN should be ≥8 to ensure RNA integrity. Please click here to view a larger version of this figure.
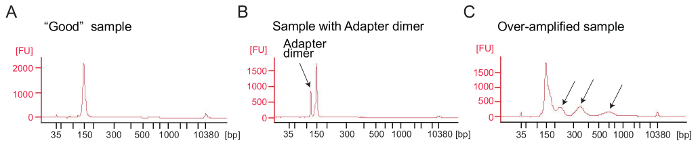
Figure 4: Bioanalyzer profiles of ribosome profiling libraries
(A) A good library: The sample shows a peak at the expected size range (140 – 160 bp) and no further purification is needed. (B) This sample shows excessive adapter dimer amplified product (120 bp) relative to the desired product (140 – 160 bp). This library requires further purification. (C) An over-amplified sample: Higher-than-expected molecular weight peaks and smeared PCR amplicons are visible (indicated by arrows). Please click here to view a larger version of this figure.
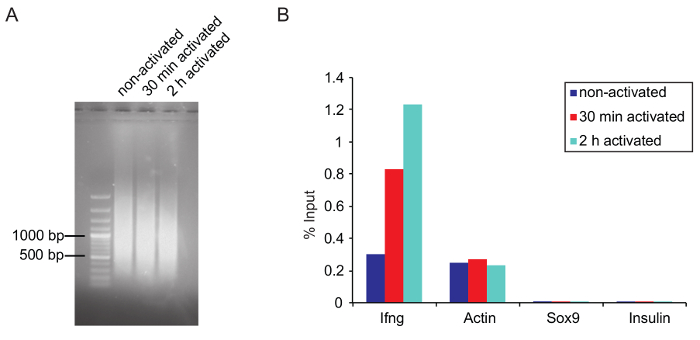
Figure 5: Optimal chromatin size after shearing and verification of ChIP by qPCR
(A) Agarose gel picture shows the optimal fragment size of the sheared chromatin from three samples that were sheared for 25 cycles on a sonicator and purified as described before in the protocol. (B) Q-PCR results of a total RNAPII ChIP (anti-RNA Pol II, 8WG16, ab817) is represented as a percentage of input. Ifng and actin primers were used as a positive while Sox9 and Insulin are negative controls (both genes are not expressed in activated Th1 cells). Please click here to view a larger version of this figure.
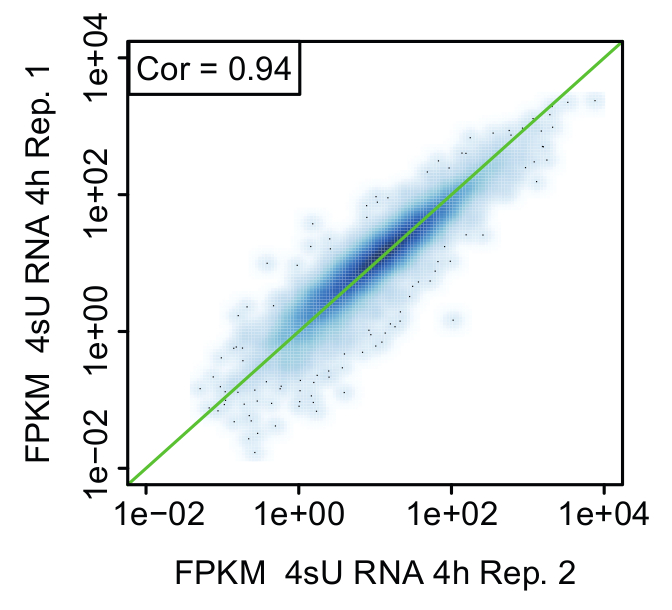
Figure 6: Comparison of biological replicates (Figure from Davari et al. 11)
Representative scatter plot comparing expression values (FPKM) between replicates of newly transcribed (4sU) RNA 4 h after stimulation of activated Th1 cells. The green line indicates equal FPKM values and rank correlation is indicated in each plot.
| Duration of labeling (min) | Recommended 4sU concentration (µM) |
| 120 | 100 – 200 |
| 60 | 200 – 500 |
| 15 – 30 | 500 – 1000 |
| <10 | 500 – 2000 |
Table 1: Recommended 4sU concentrations (from Rädle, et al.19)
The range of recommended 4sU concentrations is indicated for different times of labeling.
| Method | Cell number | RNA amount |
| 4sU-labeling | ≥2 x 107 | ≥60µg |
| Ribosome profiling | ≥2 x107 | |
| ChIP-seq | ≥2 x 107 - 3 x 107 |
Table 2: Required amount of primary T cells
Minimum amount of required primary T cells for each method. Amounts can be less when using other cell types.
