The most technically challenging step is the manual isolation, which aims to achieve a high purity of a differentiated P0 hPSC-RPE population. After a successful isolation, >90% cells in the P0 population will grow and mature to display signature RPE morphologies (Figure 1C). The existence of a minor portion of non-RPE or immature RPE cells in the P0 population is almost inevitable, but will not interfere with the downstream experiments as long as the number of pigmented and cobblestone-shaped hPSC-RPE cells is the overwhelming majority.
With the hPSC-RPE based disease-in-a-dish approach, each patient-specific BEST1 mutation can be comprehensively characterized in vivo for its protein expression (immunoblotting, Figure 3), membrane trafficking (immunostaining, Figure 4), and ion channel function (whole-cell patch clamp, Figure 5). These results will provide critical information for elucidating the pathological mechanisms of the channel mutations, and for developing personalized medicine.
As BEST1 is predominantly expressed in RPE cells, it can be used as a cellular marker for the validation of a mature RPE status. It should be noted that some BEST1 mutations might affect its protein expression, so other well-established RPE markers such as RPE65 and CRALBP still need to be evaluated (Figure 3).
Matured P0 hPSC-RPE cells can be maintained for 2-3 months before splitting (to P1) for whole-cell patch clamp. P0 cells older than 3 months are not recommended for patch clamp but can still be used for immunoblotting and immunostaining experiments.
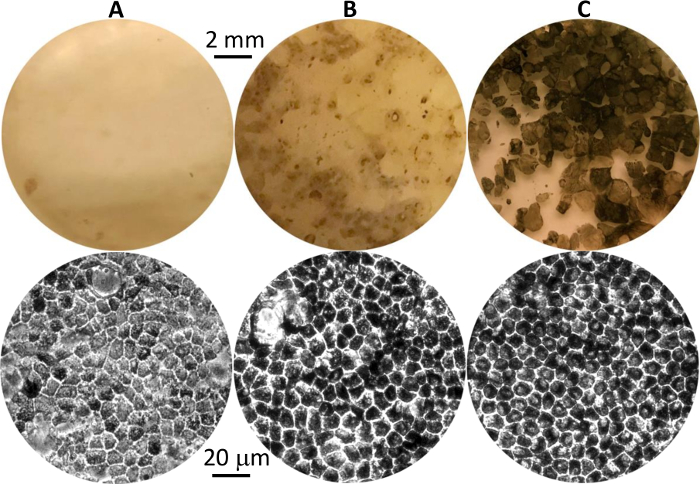
Figure 1: Differentiated hPSC-RPE cells at different stages. Representative images of pigmented hPSC-RPE clusters in culture plates at first appearance (A), before isolation (B), and after growth to maturity post isolation (C). Eye-view and 20X phase contrast images are shown in the top and bottom panels, respectively. Please click here to view a larger version of this figure.

Figure 2: Representative image of the micro cell scraper pulled from a Pasteur pipette. Please click here to view a larger version of this figure.
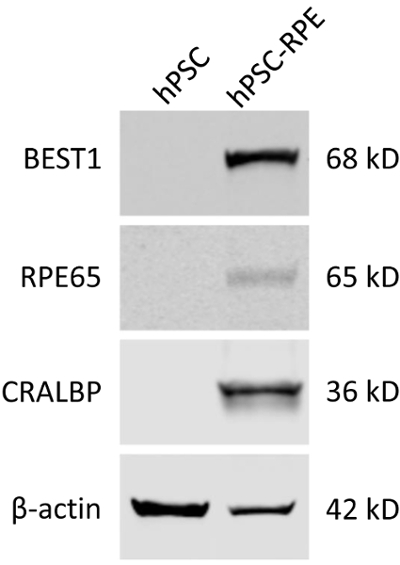
Figure 3: Validation of the mature RPE status of differentiated hPSC-RPE cells by immunoblotting. Blots showing the expression of RPE-specific protein markers BEST1, RPE65, and CRALBP in WT hPSC and hPSC-RPE cells. Please click here to view a larger version of this figure.
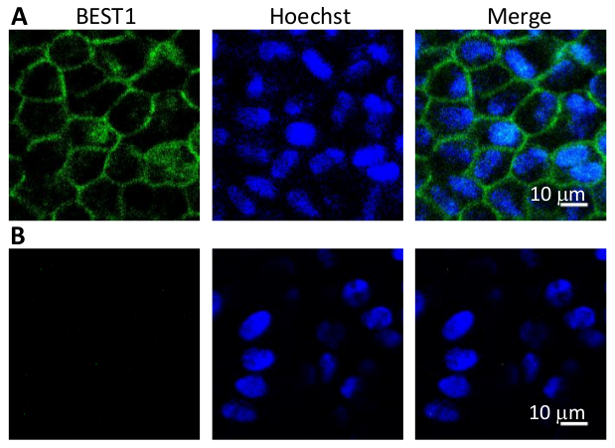
Figure 4: Immunostaining of BEST1 in WT hPSC-RPEs. (A) Representative immunofluorescent images showing the membrane localization of WT BEST1 in cobblestone-shaped hPSC-RPE cells. (B) Immunostaining negative control omitting the BEST1 primary antibody.
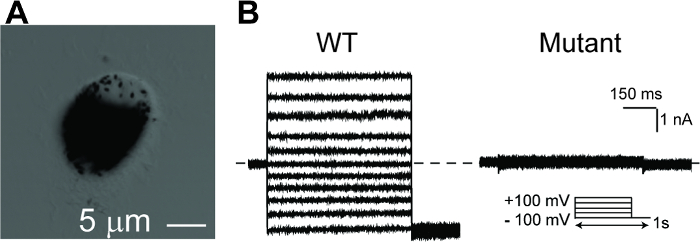
Figure 5: Whole-cell patch clamp recordings of hPSC-RPEs. (A) A single hPSC-RPE cell for whole-cell patch clamp. (B) Representative current traces recorded from a BEST1 WT hPSC-RPE and a BEST1 mutated hPSC-RPE at peak Ca2+. The voltage protocol used to elicit currents is shown in the Insert. Scale bar = 1 nA, 150 ms. See Tables 3 and Table 4 for patch clamp preparation details. Please click here to view a larger version of this figure.
| Reagent | Amount |
| Knock-Out (KO) DMEM | 500 mL |
| KO serum replacement | 15% (75 mL) |
| Nonessential amino acids | 1% (5 mL) |
| Glutamine | 1% (5 mL) |
| Penicillin-streptomycin | 1% (5 mL) |
| Nicotinamide | 10 mM |
| Human activin-A* | 100 ng/mL |
| *Human activin-A is supplemented during days 15–28 of the differentiation protocol. | |
Table 1: RPE Differentiation Medium.
| Reagent | Amount |
| MEM (α modification) | 500 mL |
| Fetal Bovine Serum | 5% (25 mL) |
| N1 supplement | 1% (5 mL) |
| Glutamine-penicillin-streptomycin | 1% (5 mL) |
| Nonessential amino acids | 1% (5 mL) |
| Taurine | 125 mg |
| Hydrocortisone | 10 µg |
| Triiodo-thyronin | 0.0065 µg |
Table 2: RPE Culture Medium.
| Reagent | Concentration |
| CsCl | 130 mM |
| MgCl2 | 1 mM |
| EGTA | 10 mM |
| Magnesium ATP | 2 mM |
| HEPES (pH 7.4) | 10 mM |
| CaCl2* | Varies |
| Glucose** | ~ 5 mM |
| *Add CaCl2 to obtain desired Ca2+ concentration | |
| ** Use glucose to adjust osmolarity to 290–295 mOsm/L | |
Table 3: Patch Clamp Internal Solution.
| Reagent | Concentration |
| NaCl | 140 mM |
| KCl | 5 mM |
| MgCl2 | 1 mM |
| CaCl2 | 2 mM |
| HEPES (pH 7.4) | 10 mM |
| Glucose* | ~ 5 mM |
| *Use glucose to adjust osmolarity to 300–305 mOsm/L. | |
Table 4: Patch Clamp External Solution.
